









 HTML
HTML

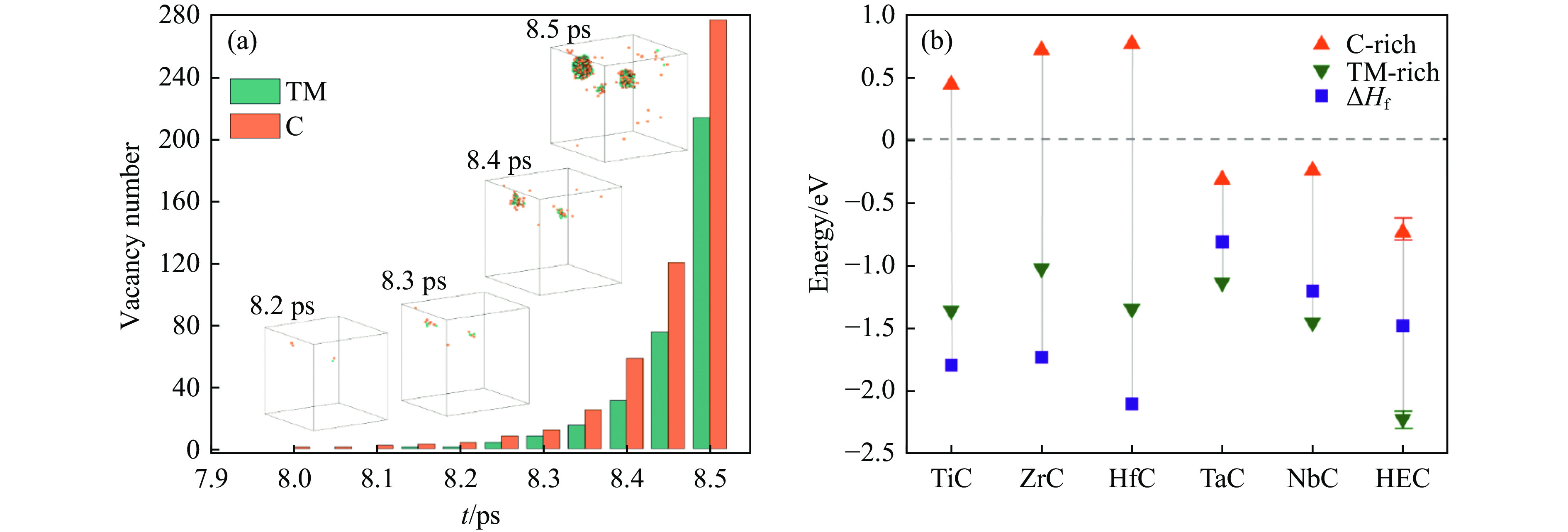
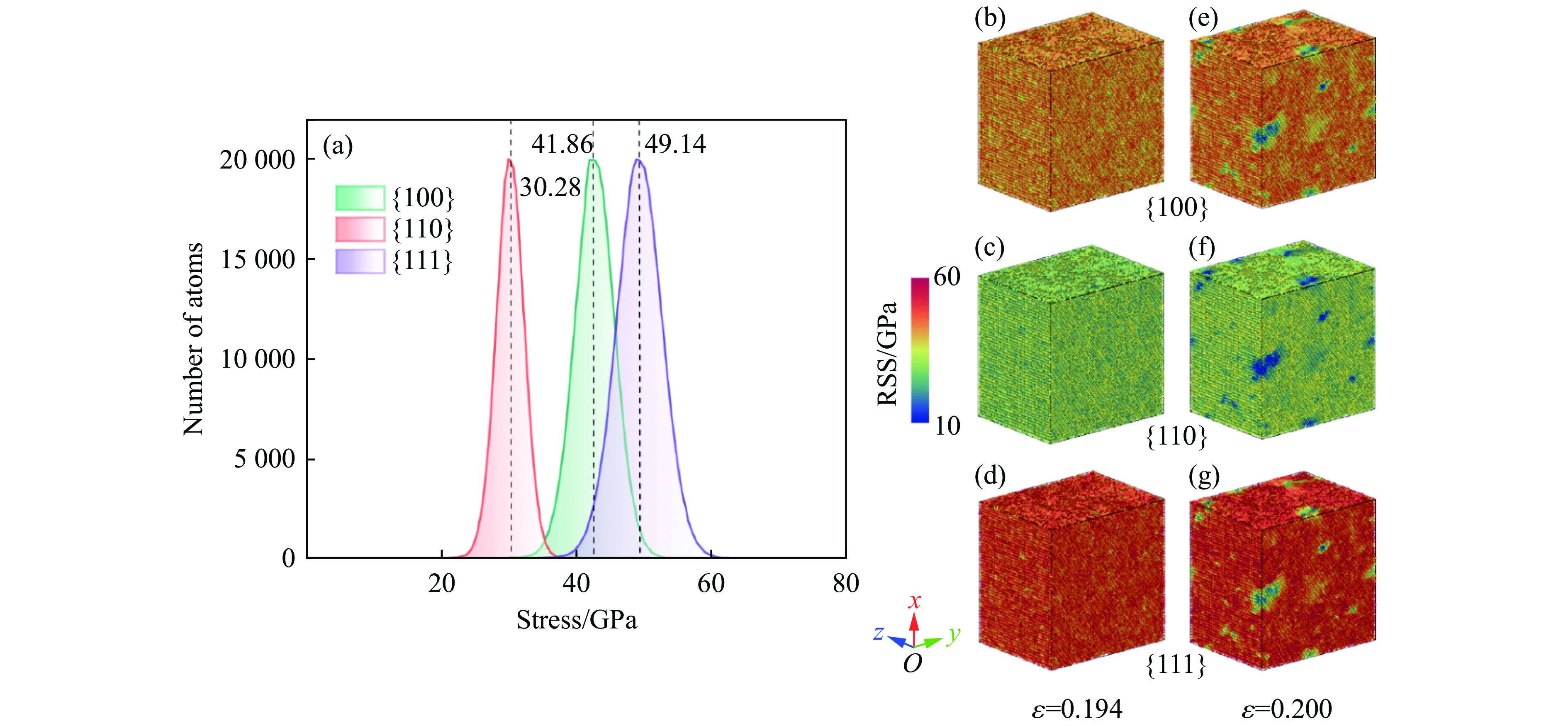
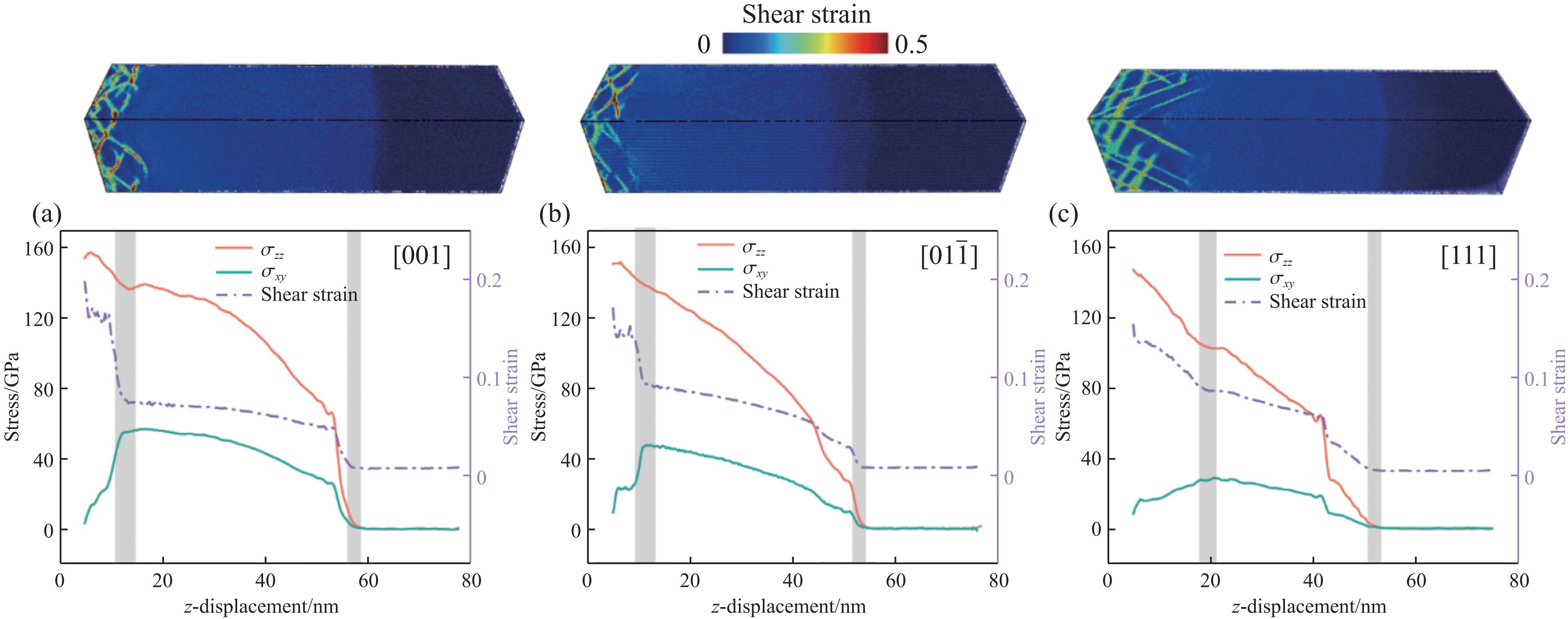
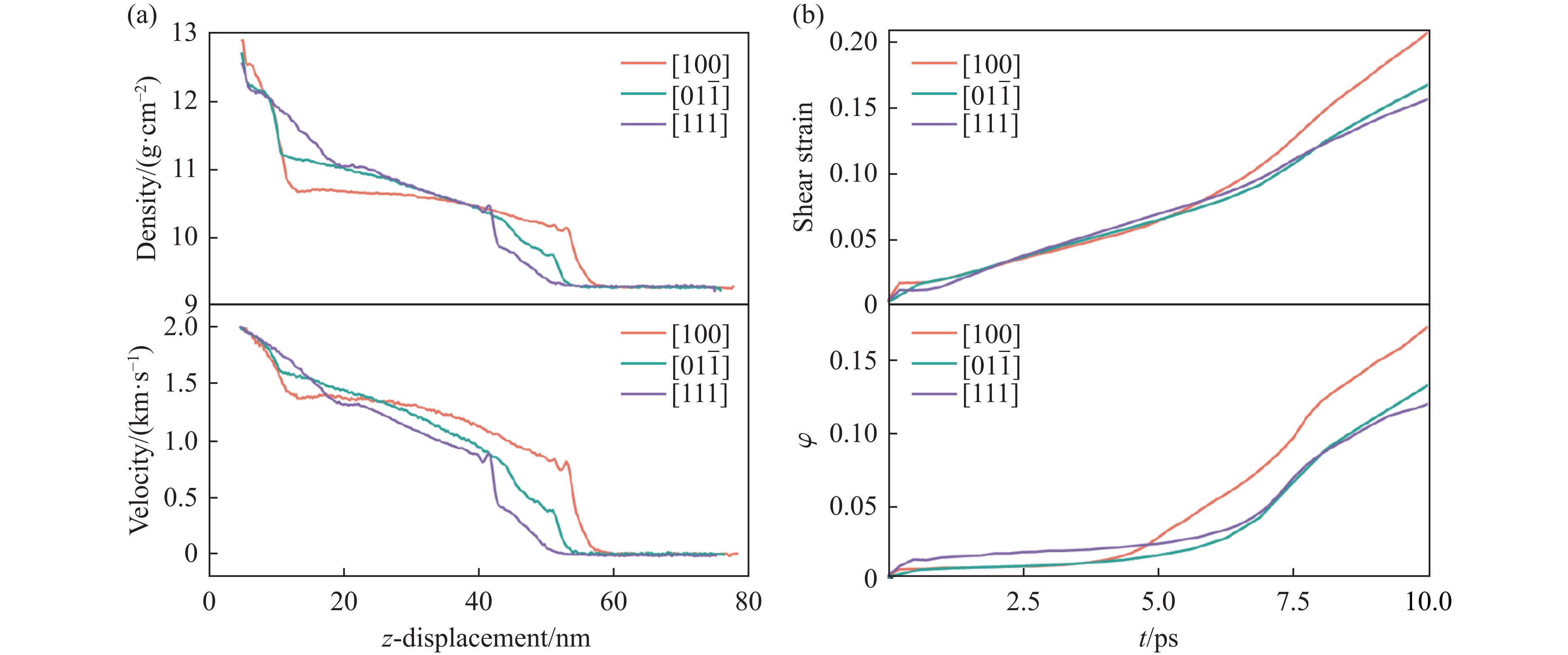

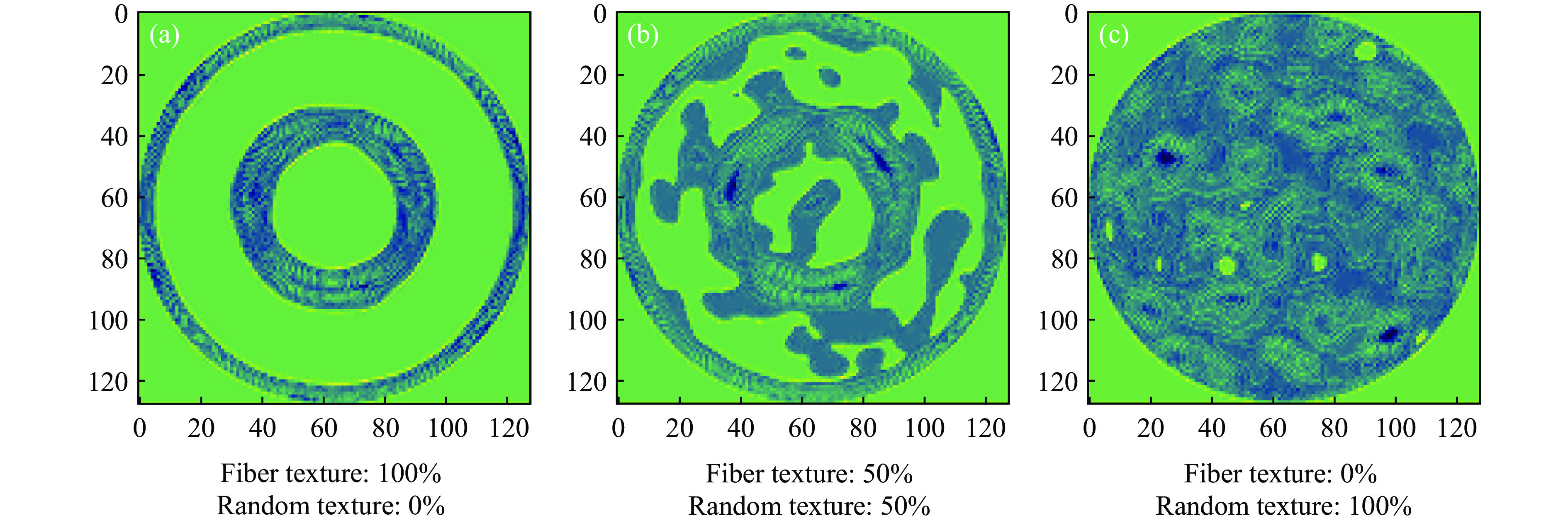
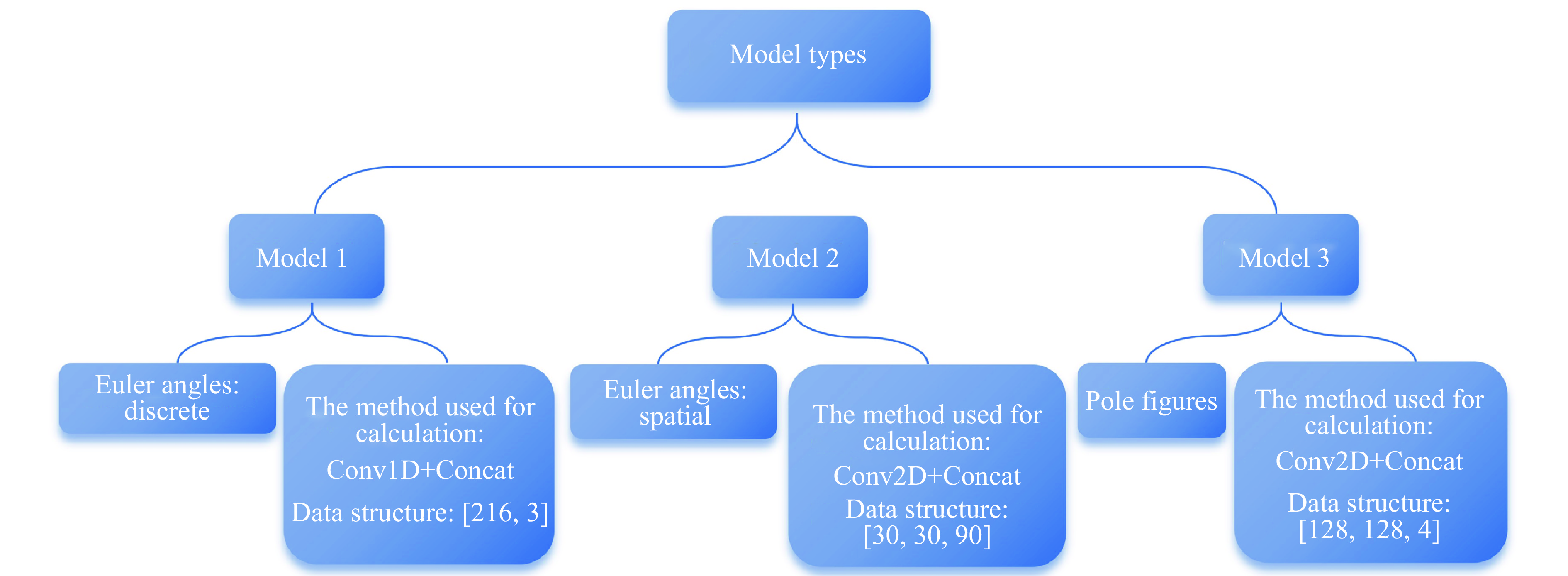
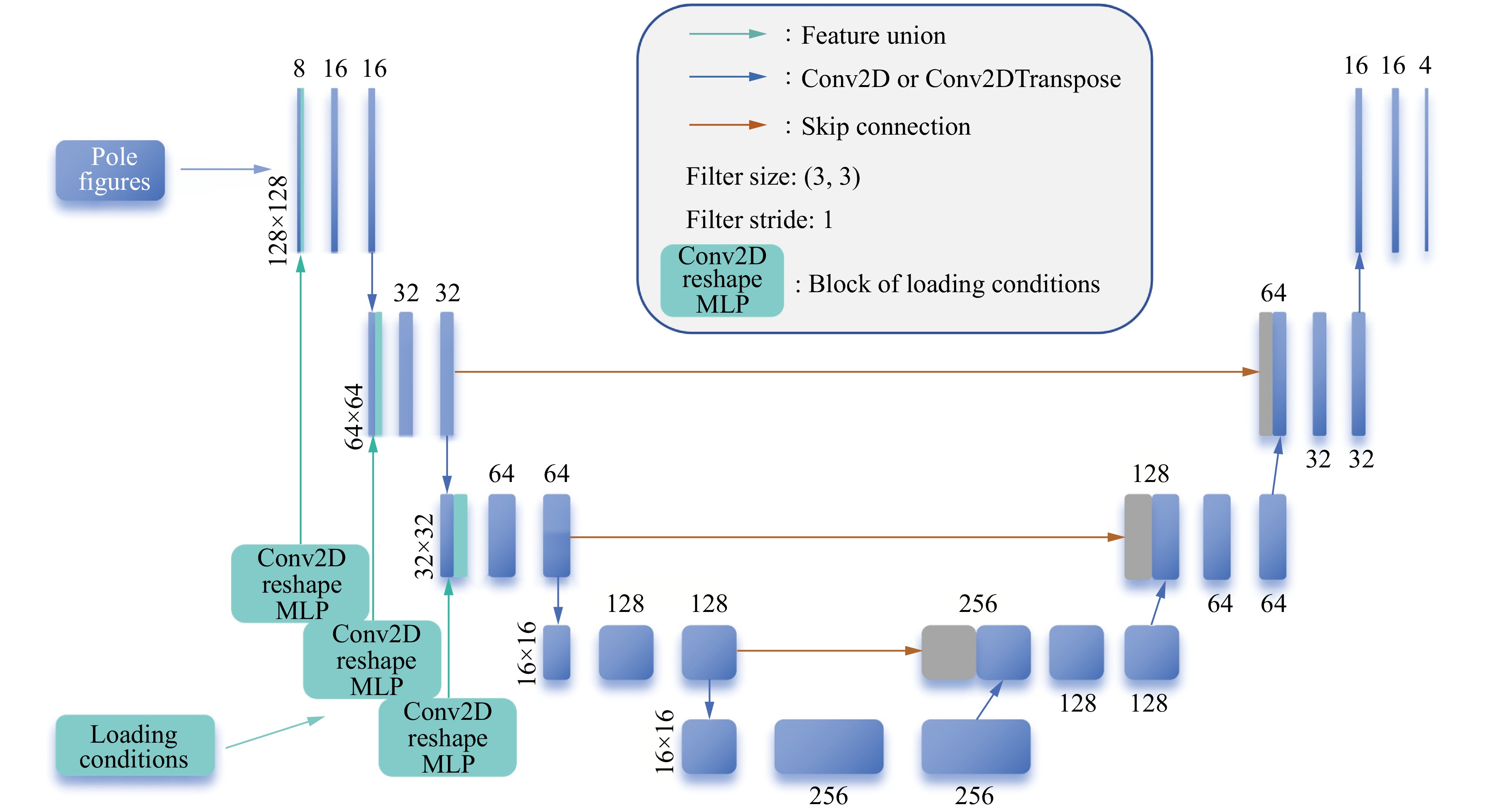
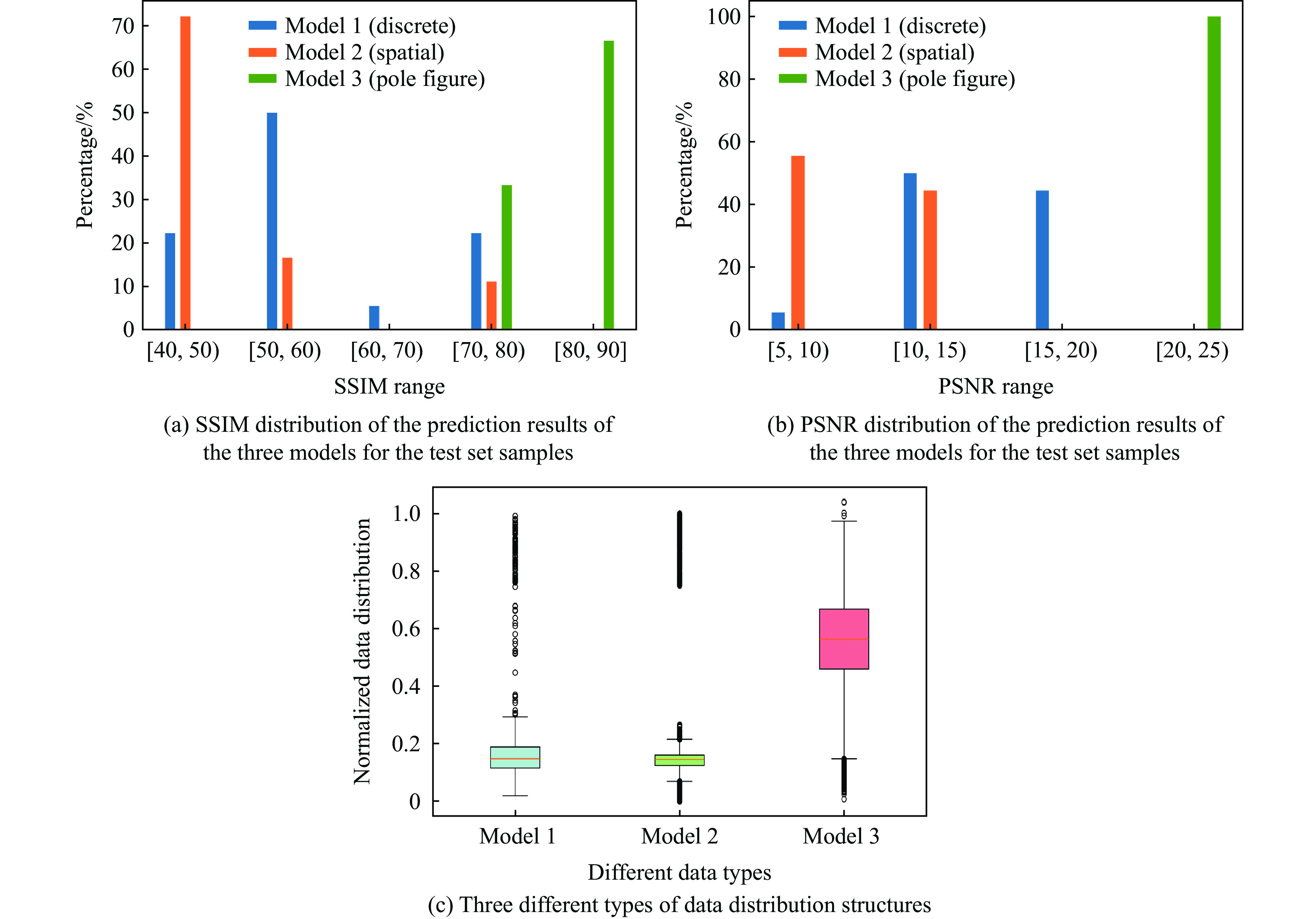

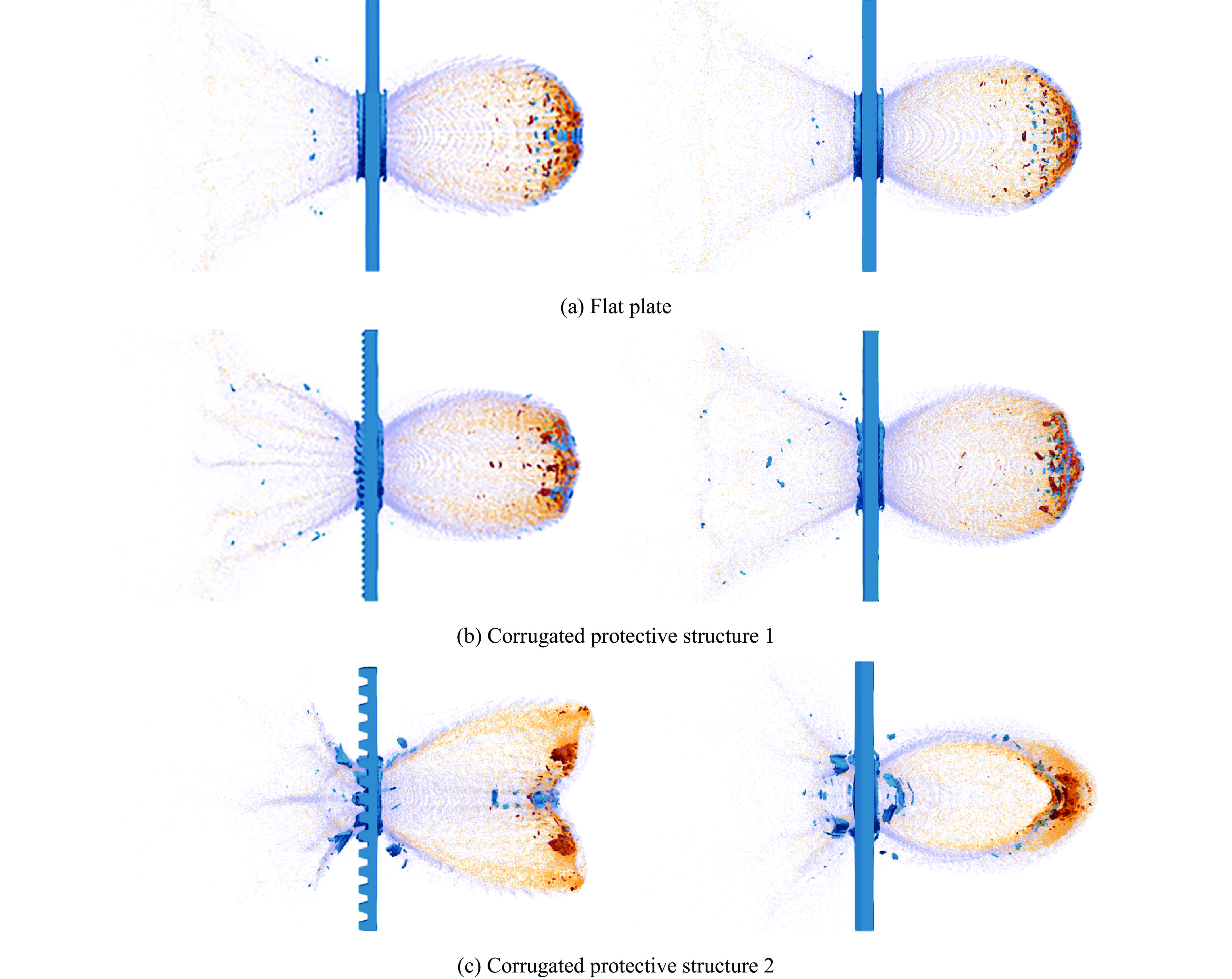
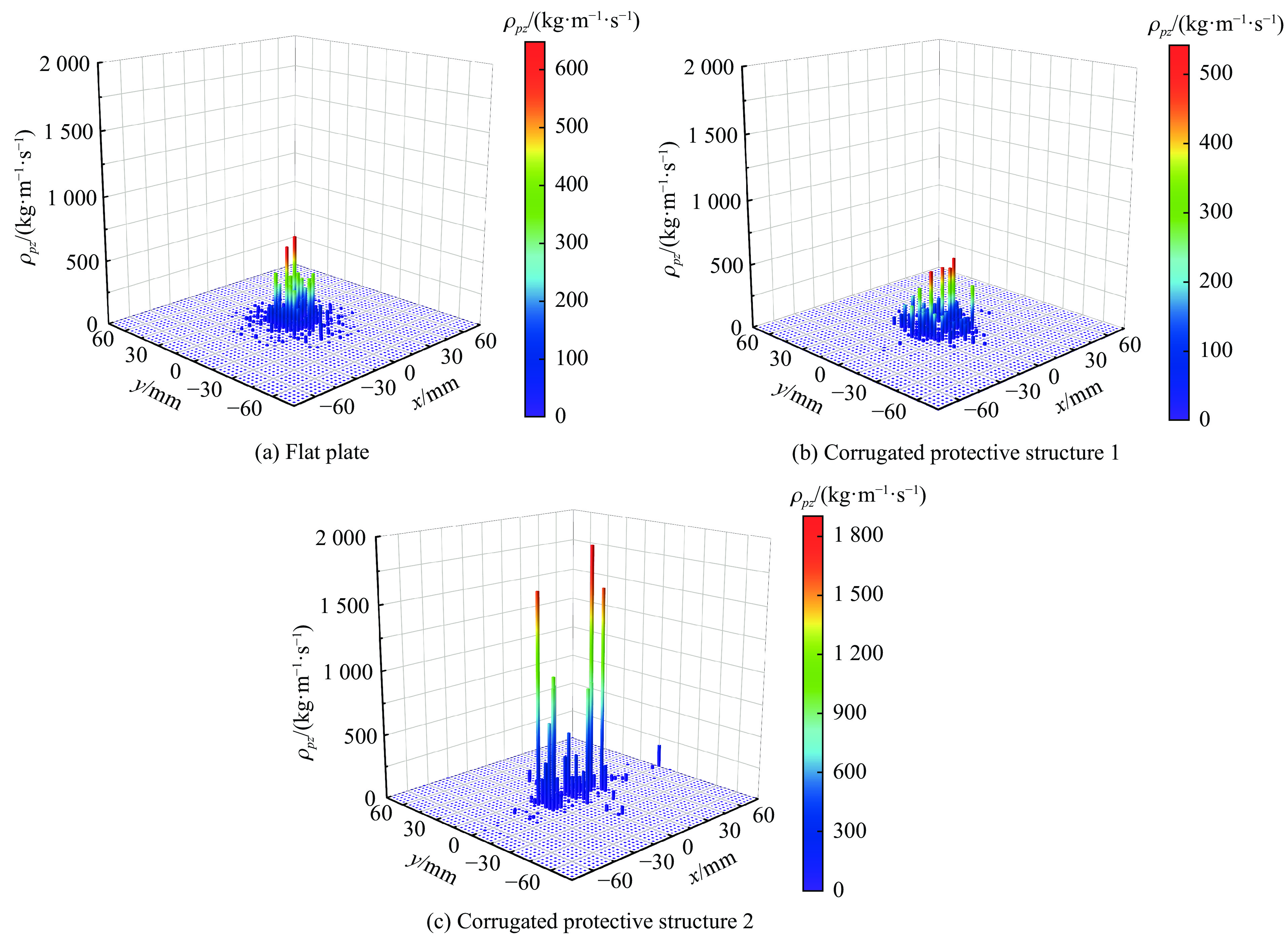
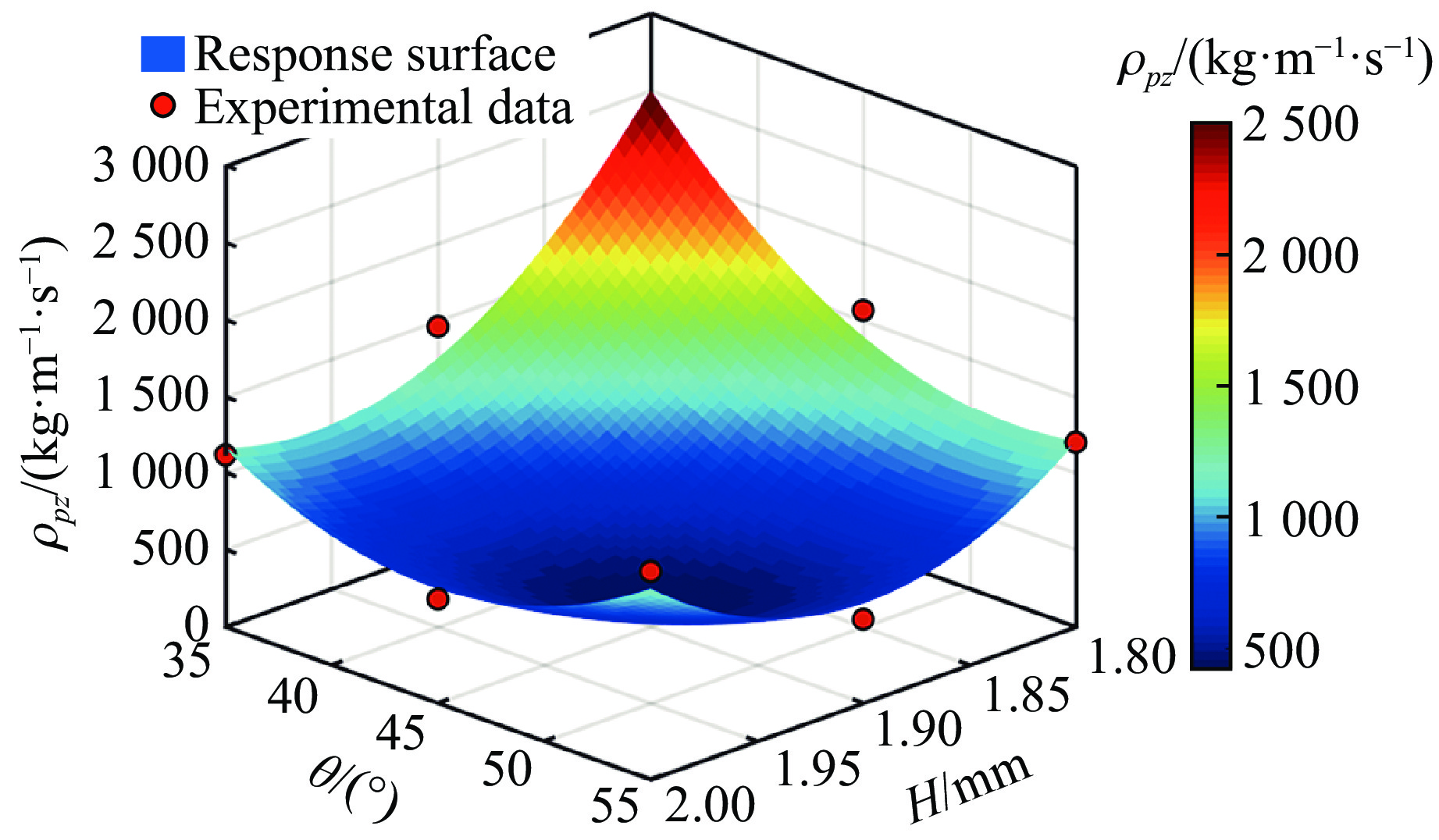
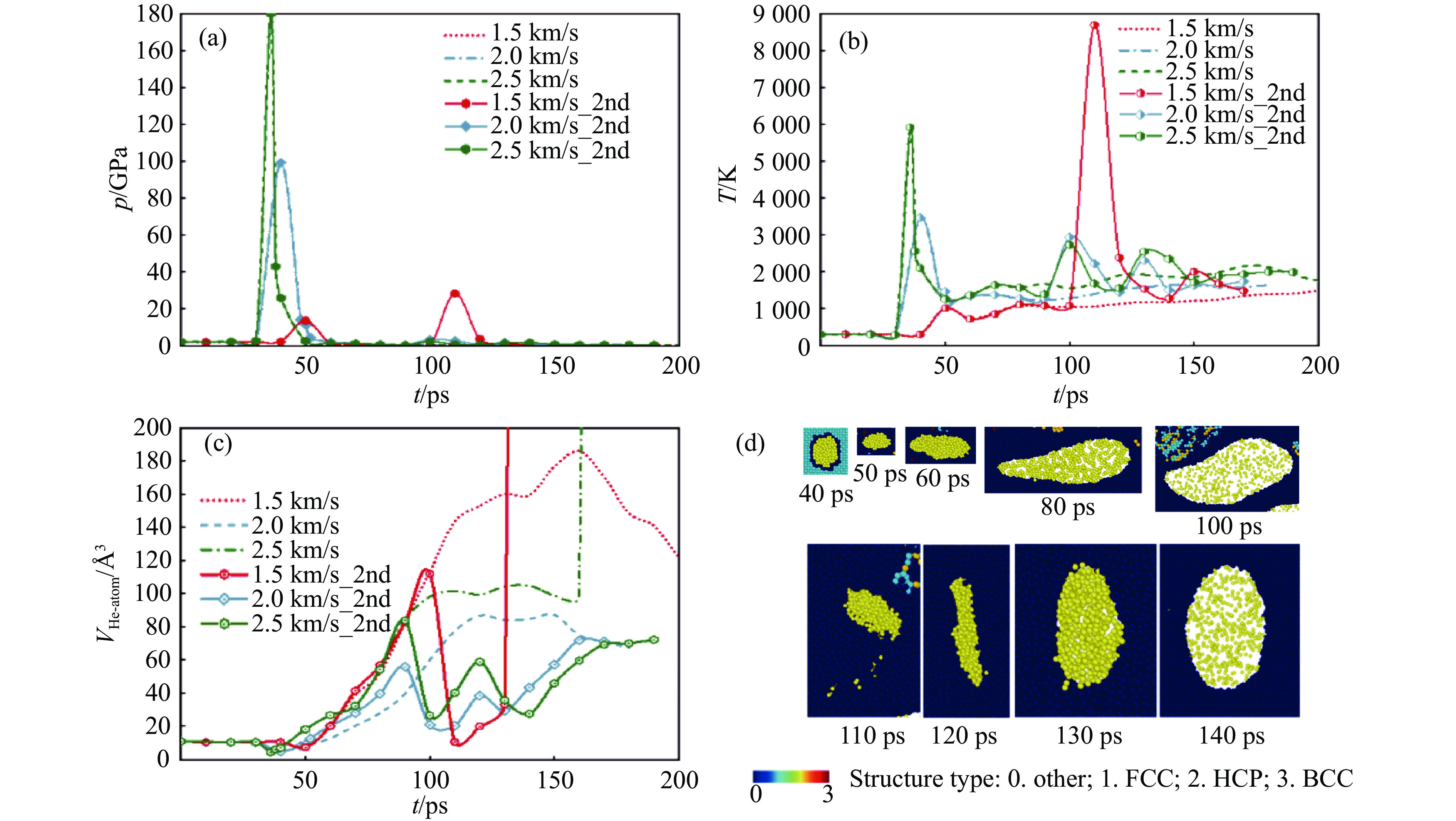
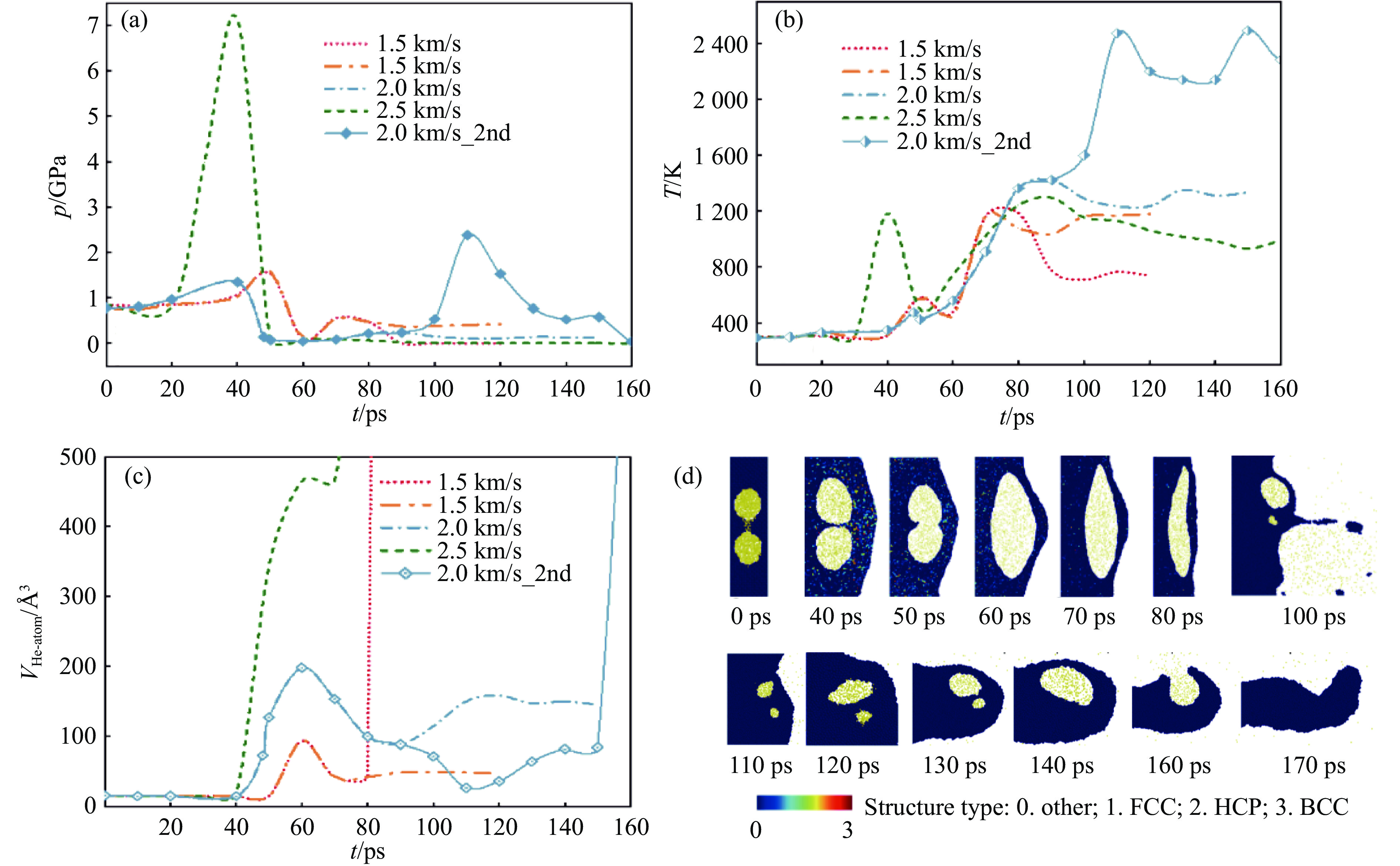
 PDF(33)
PDF(33)
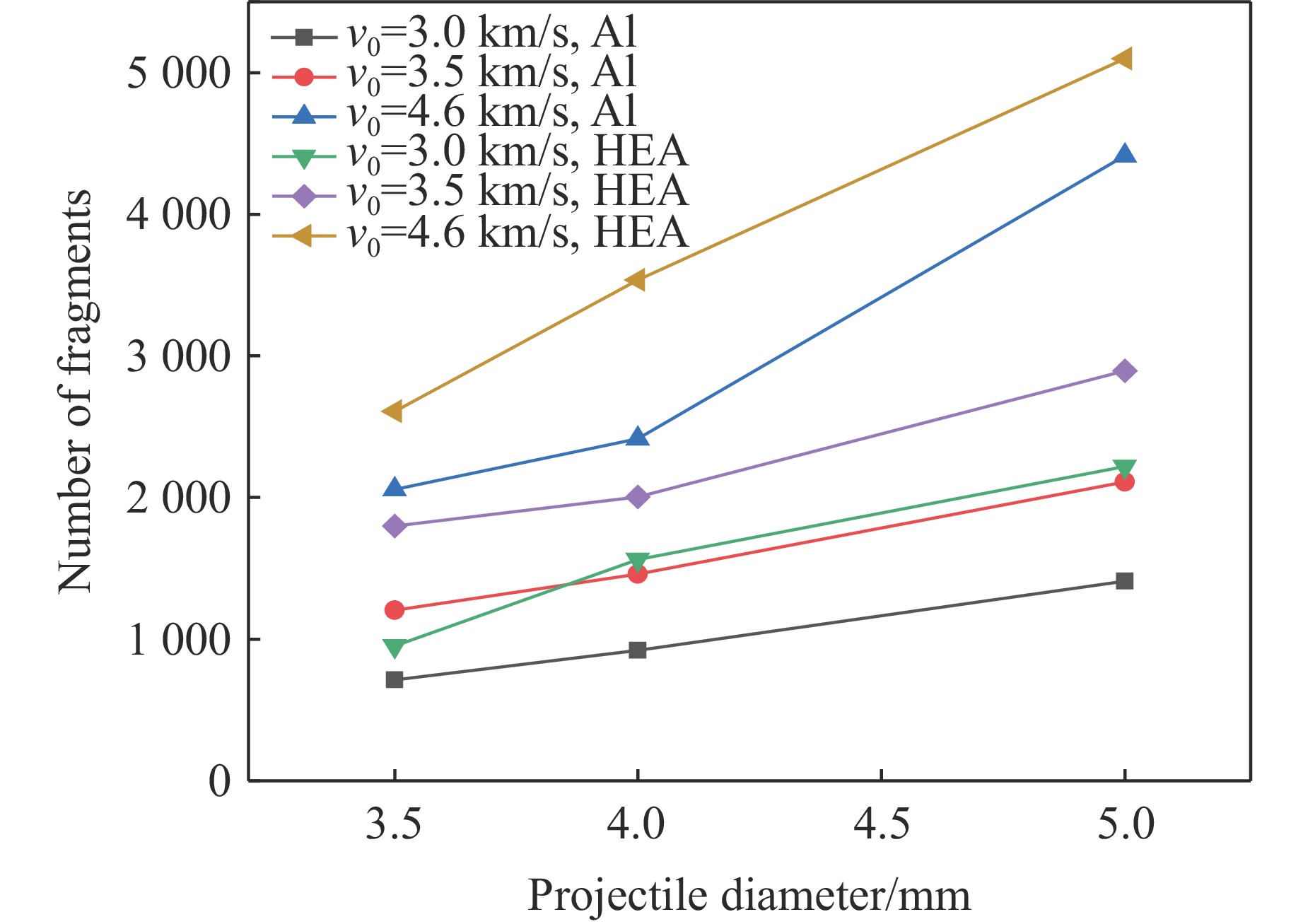
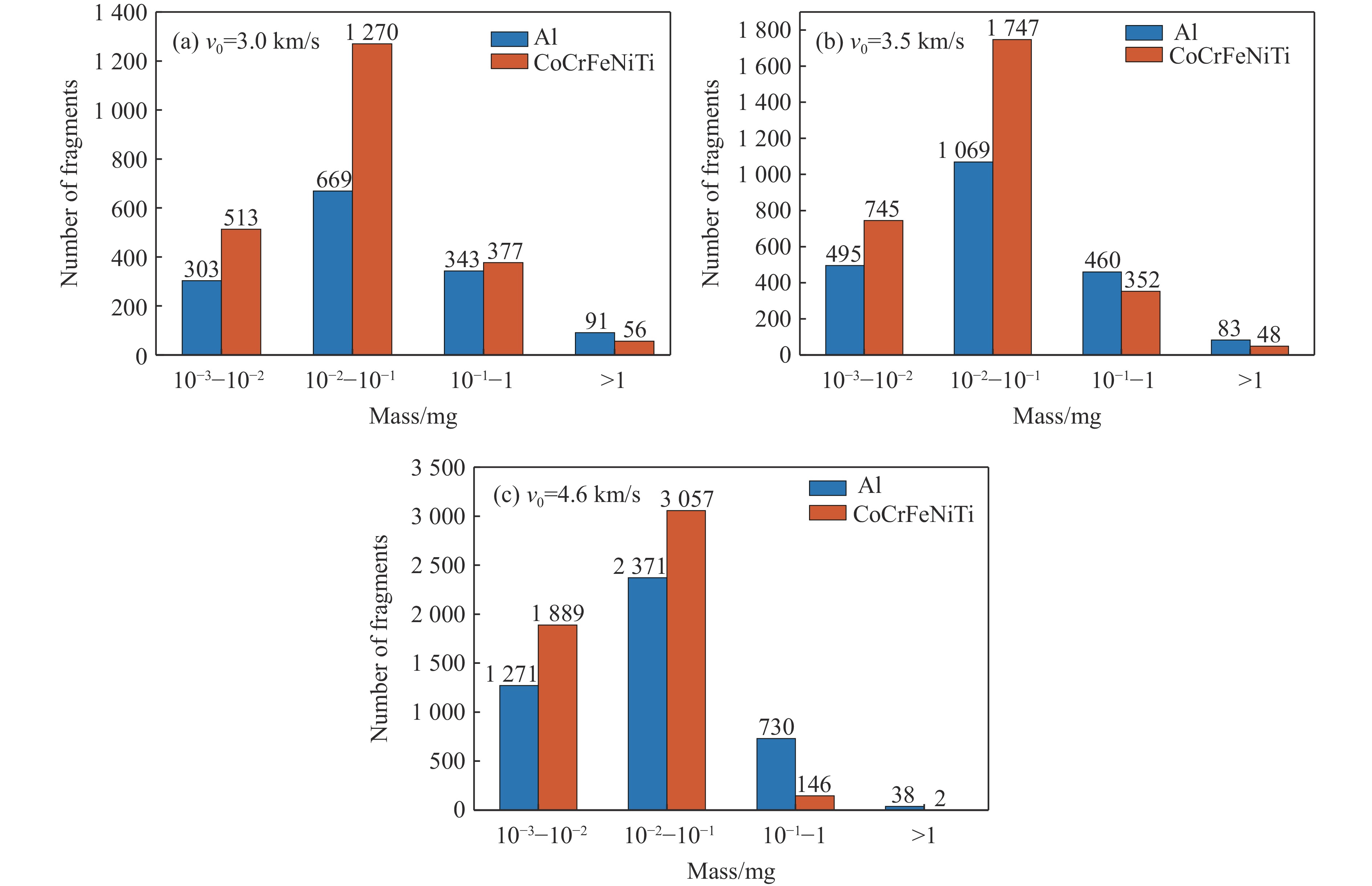

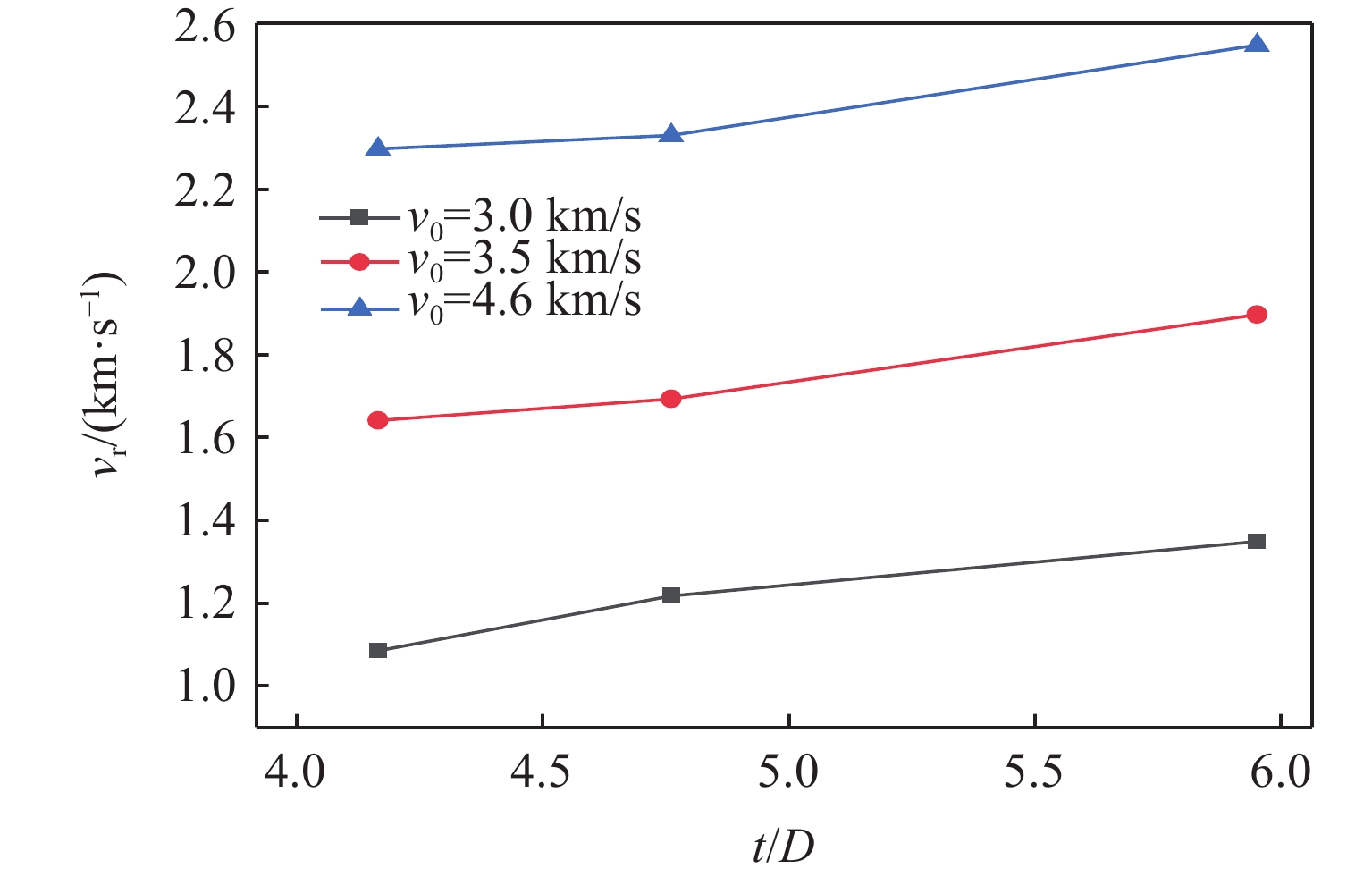
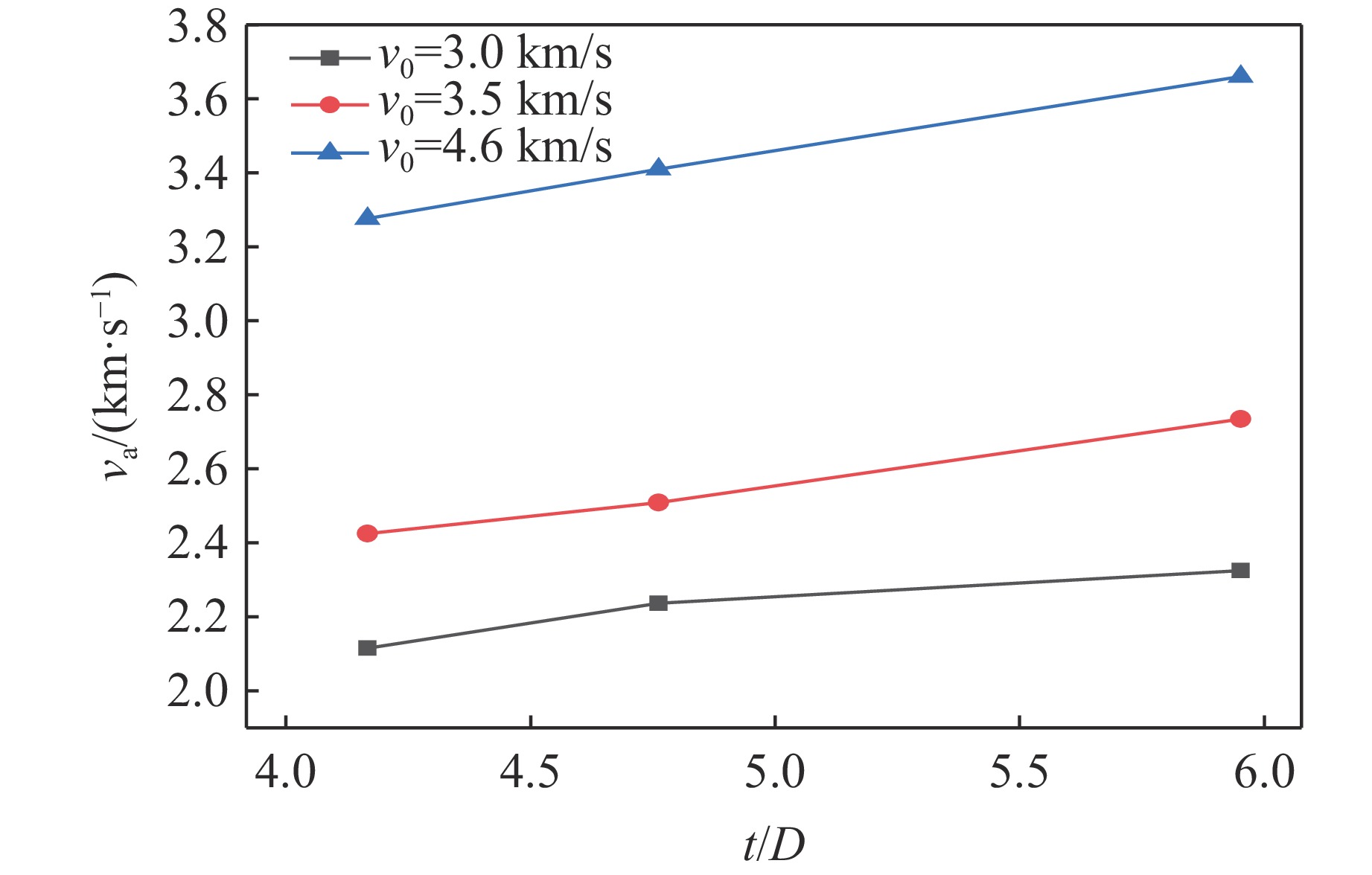
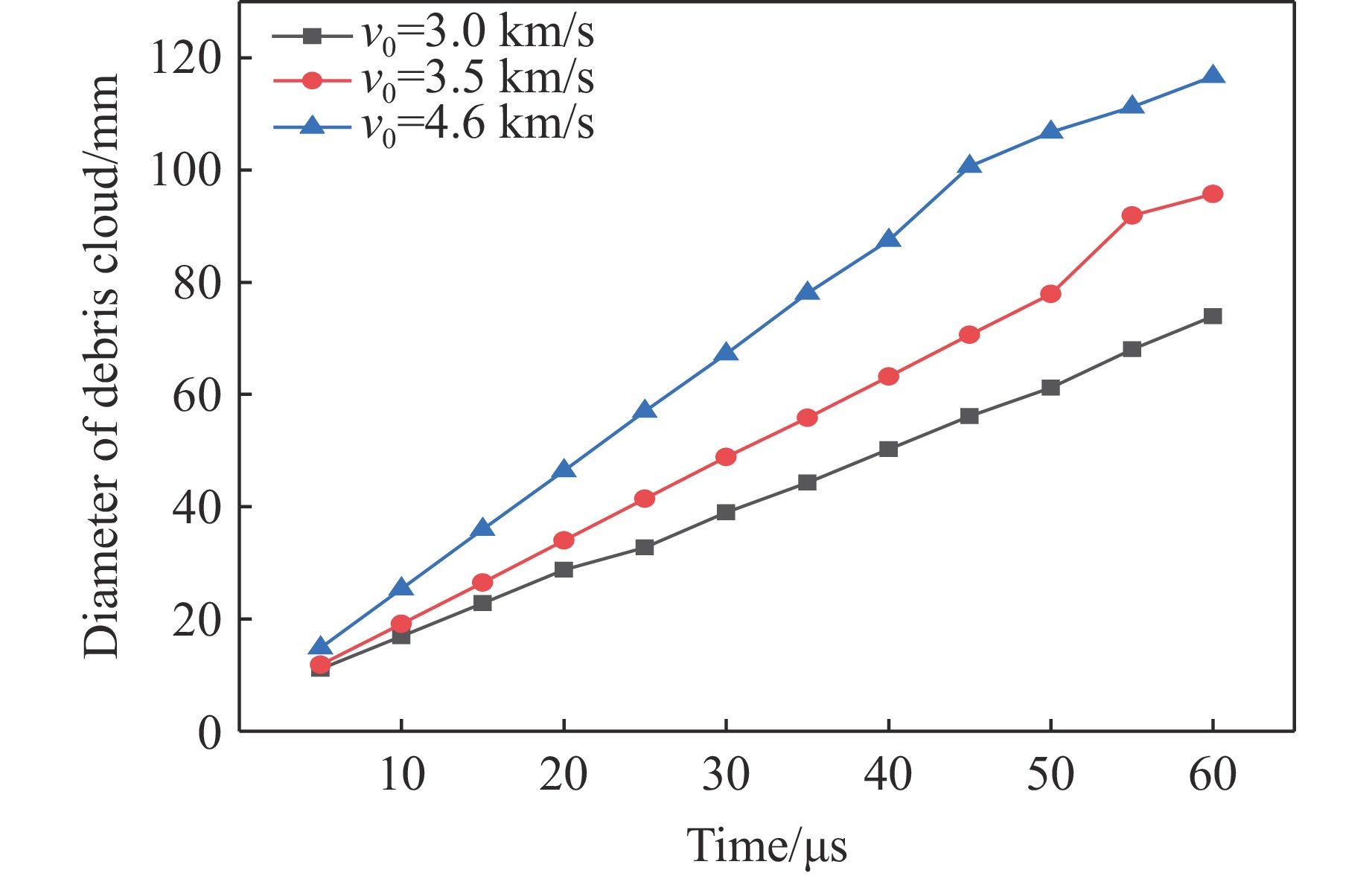
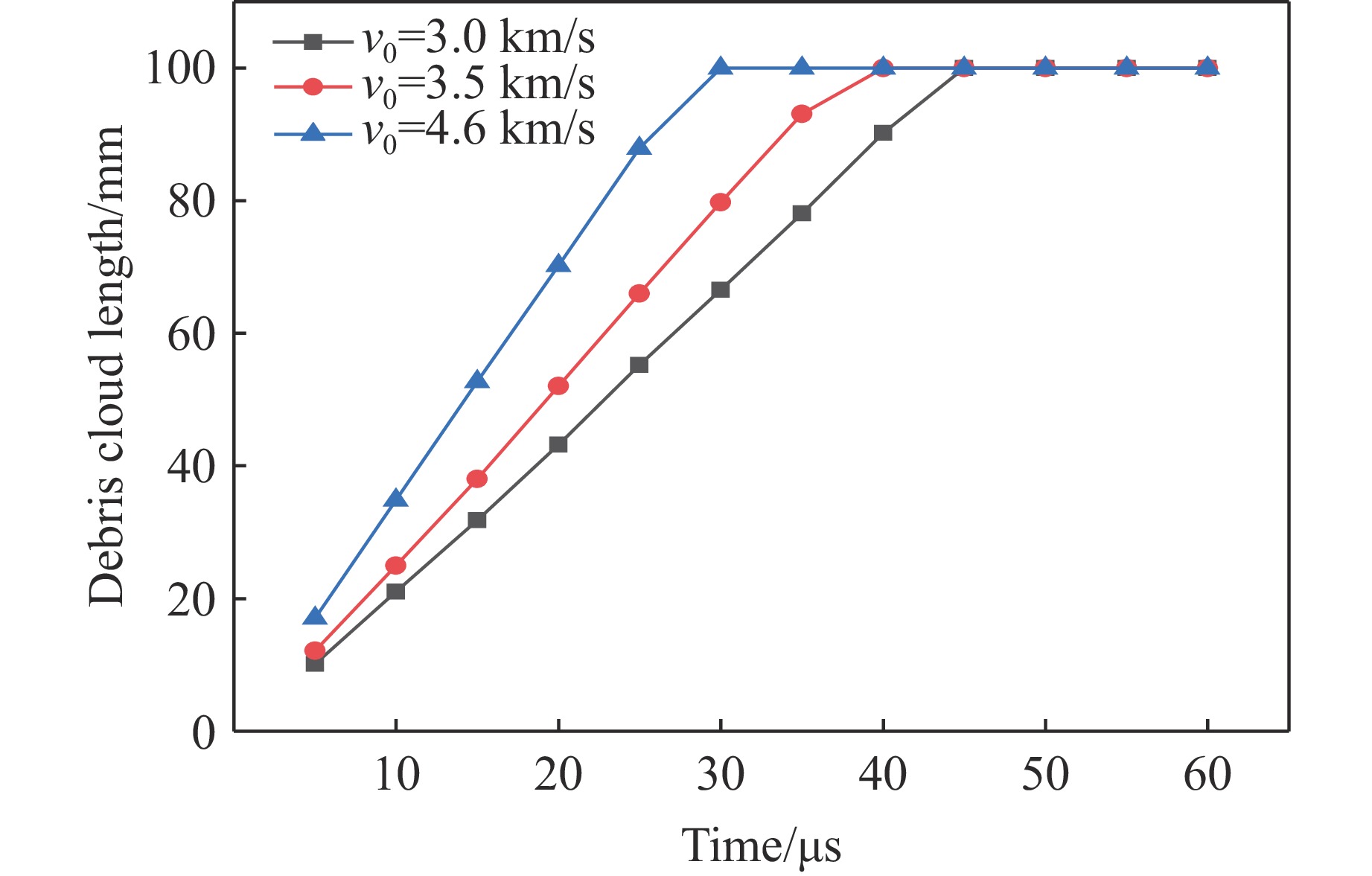
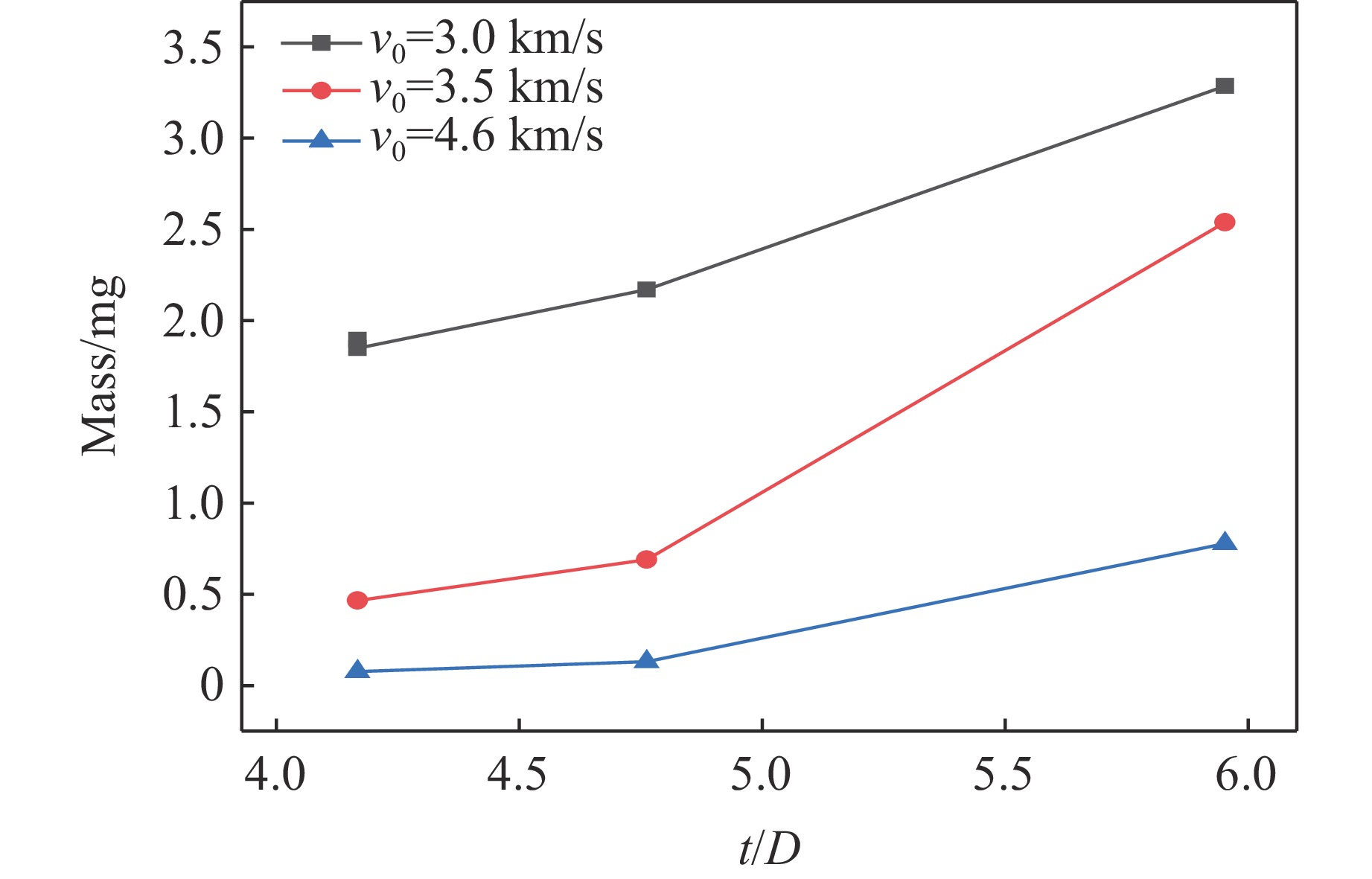
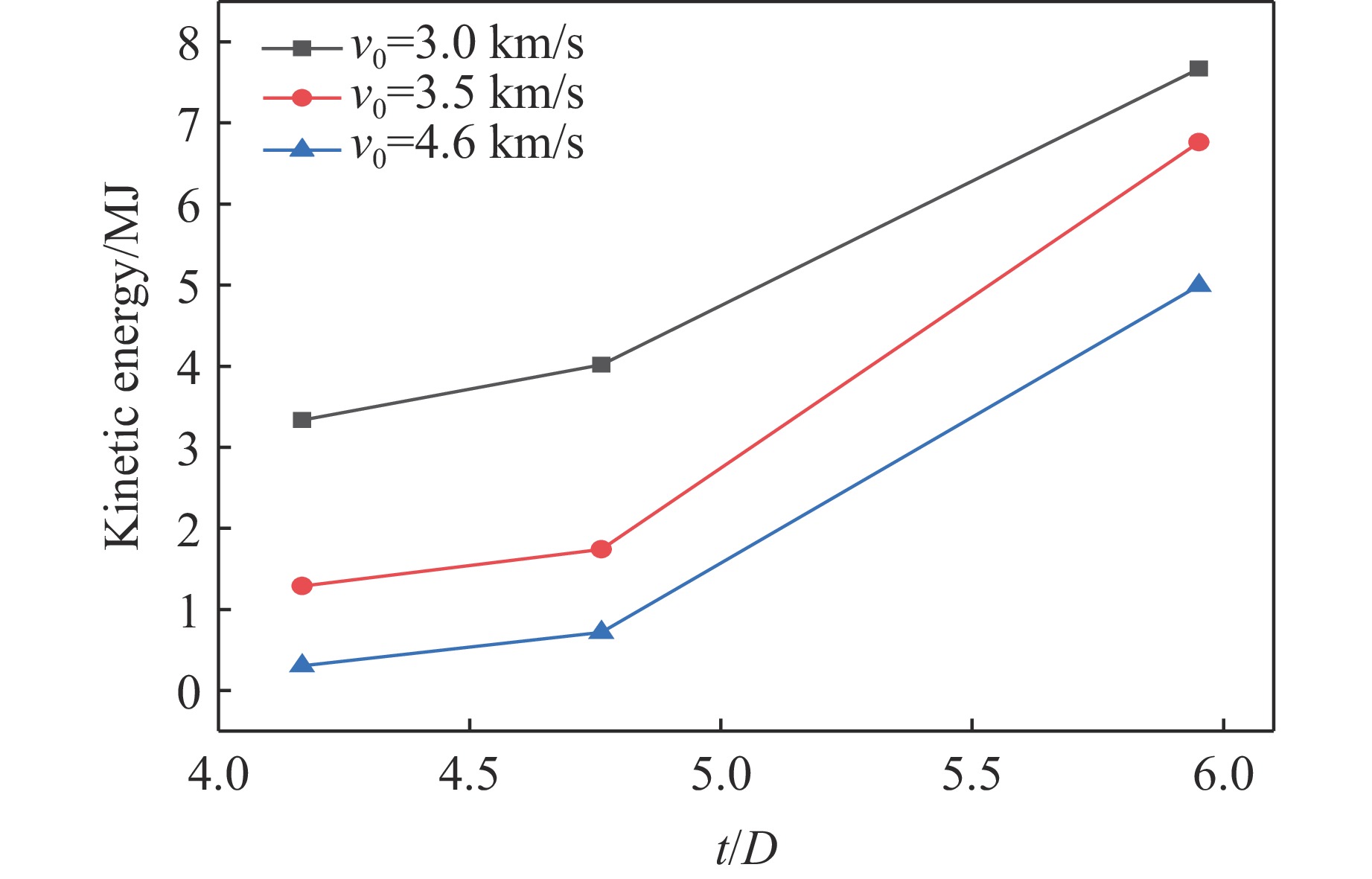
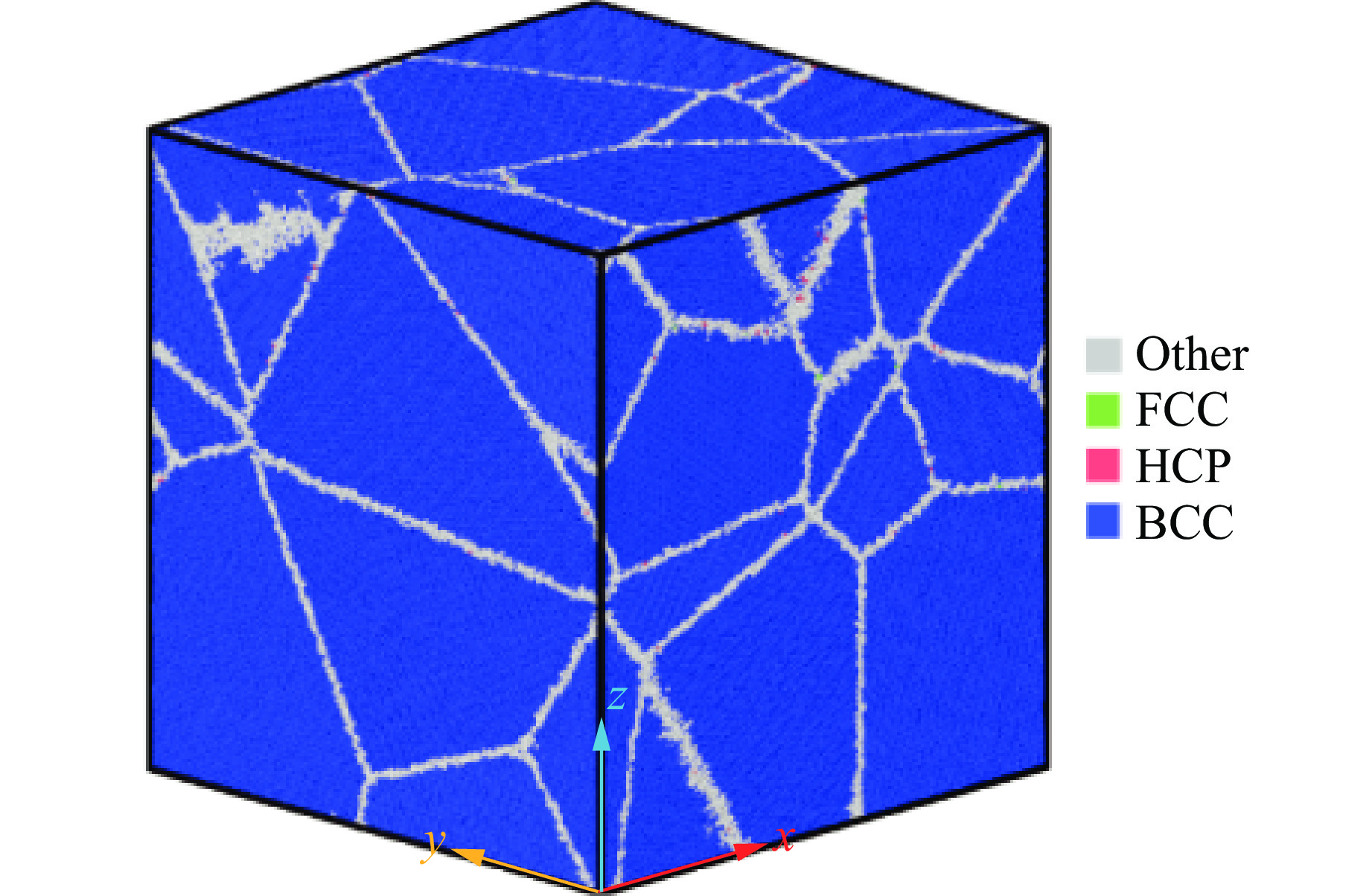
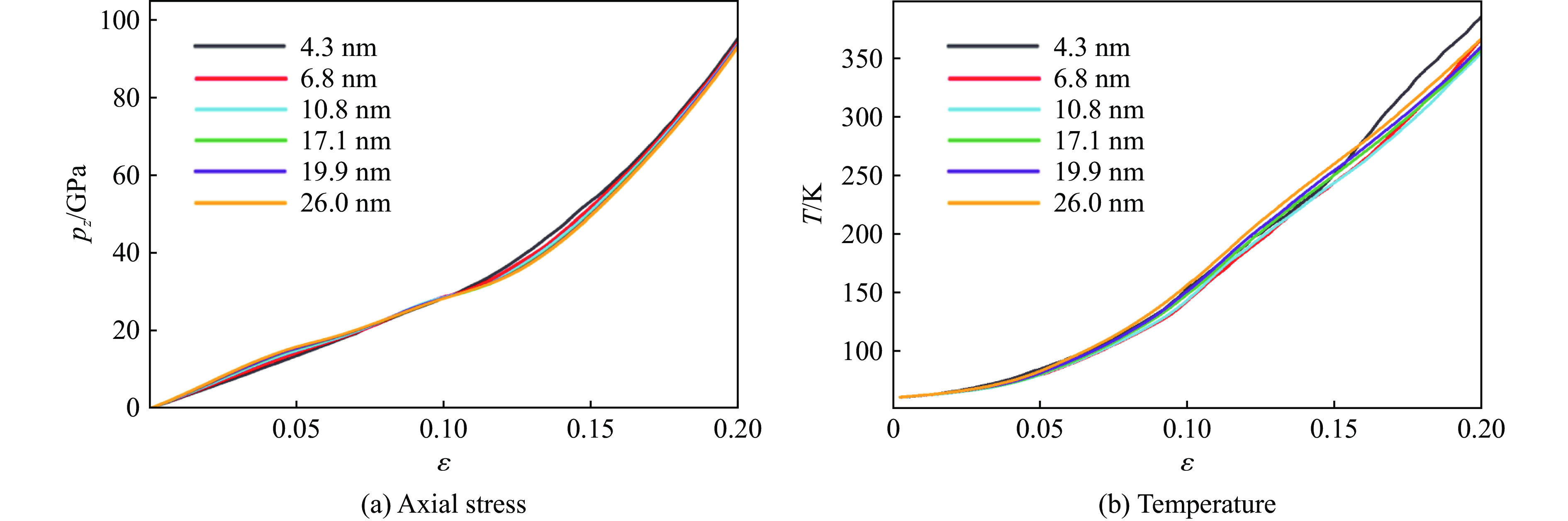
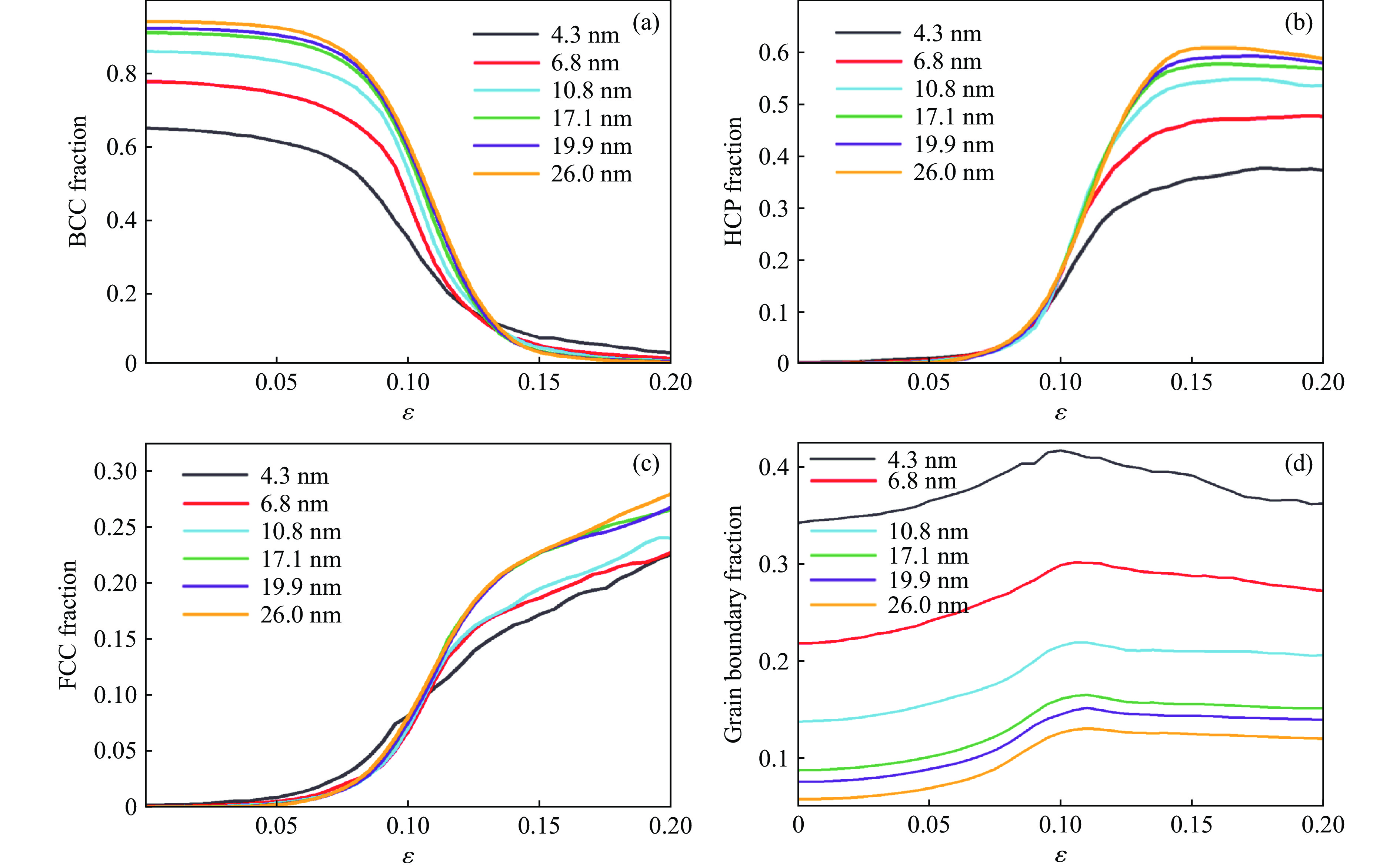
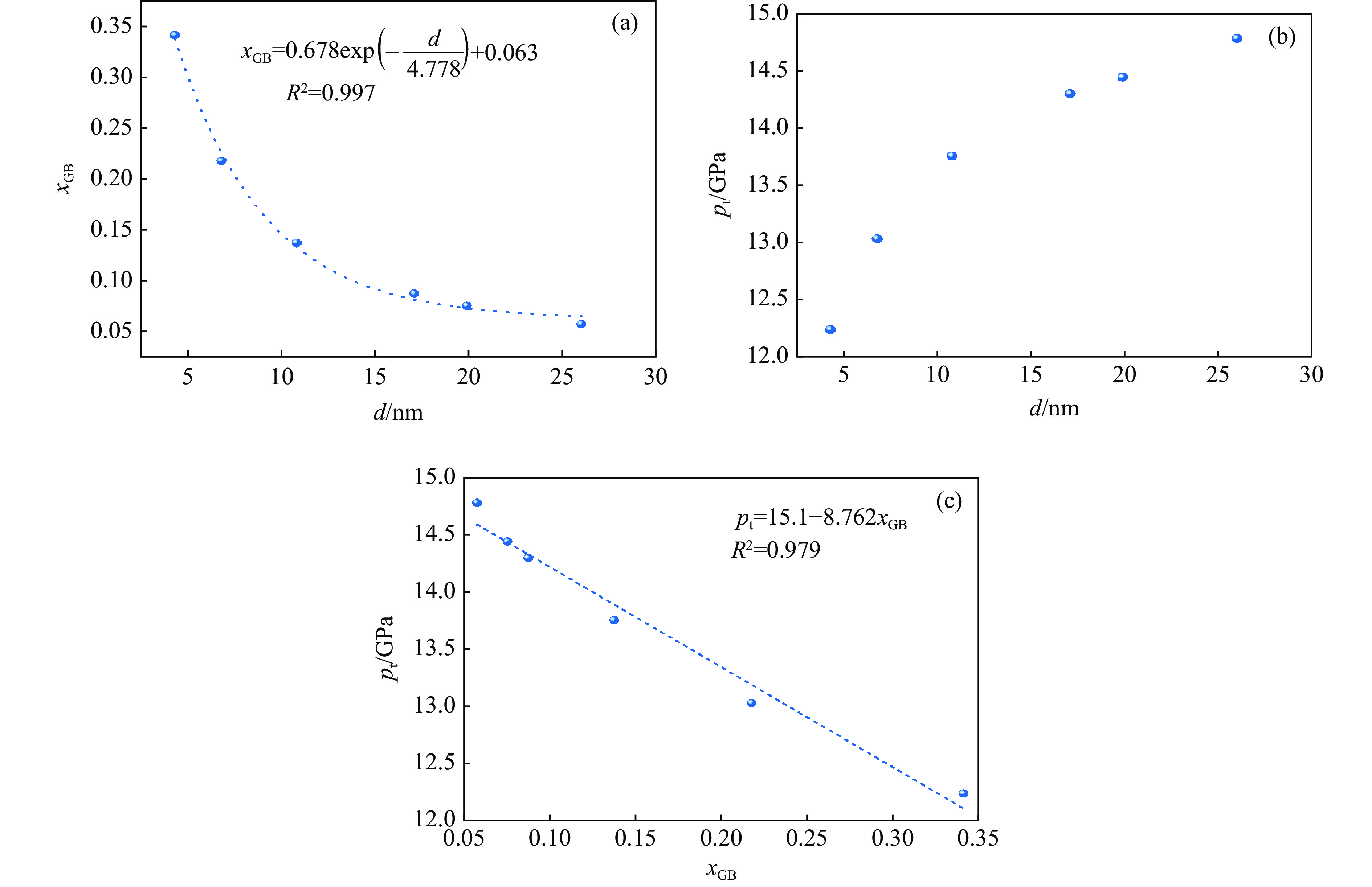
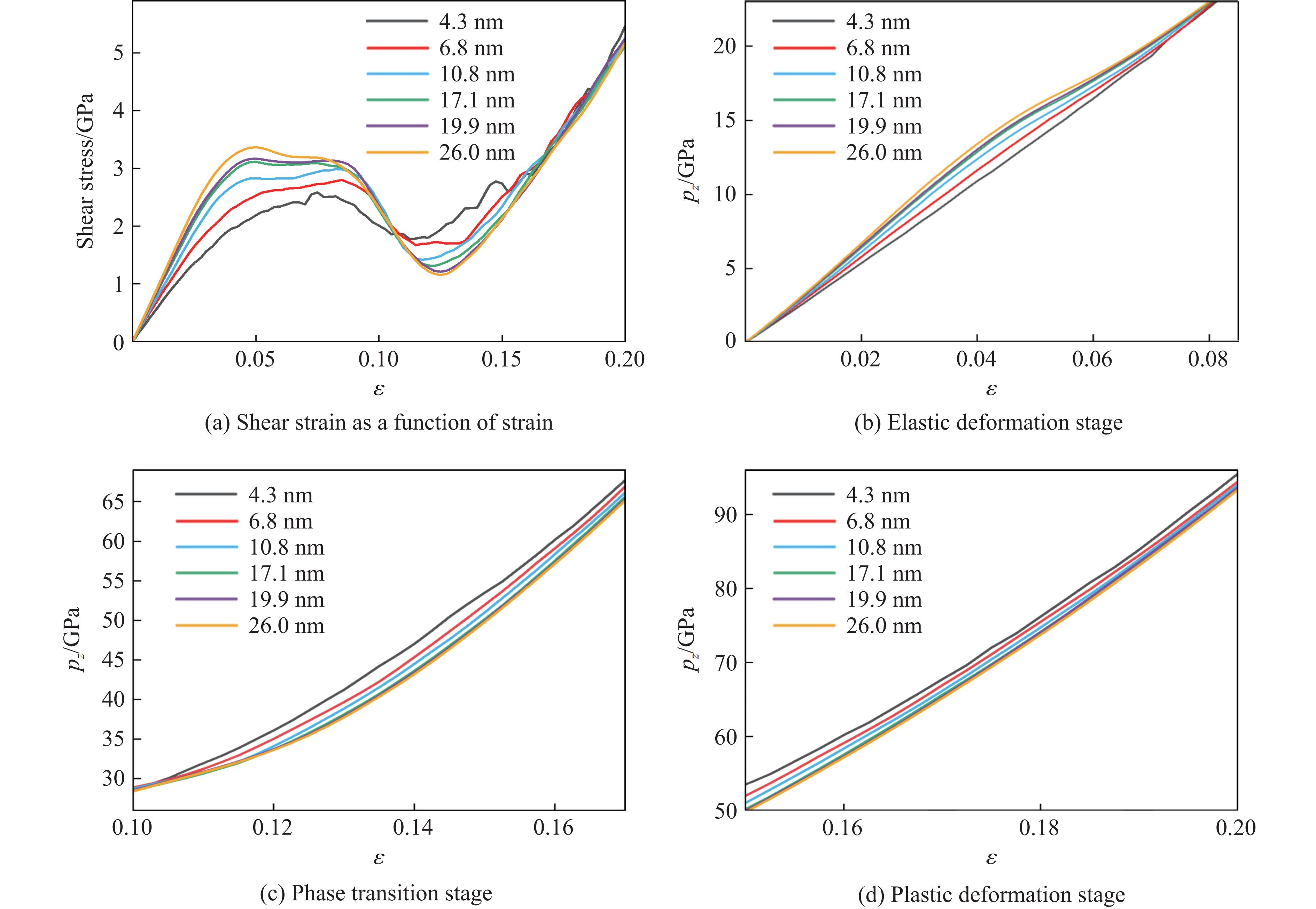
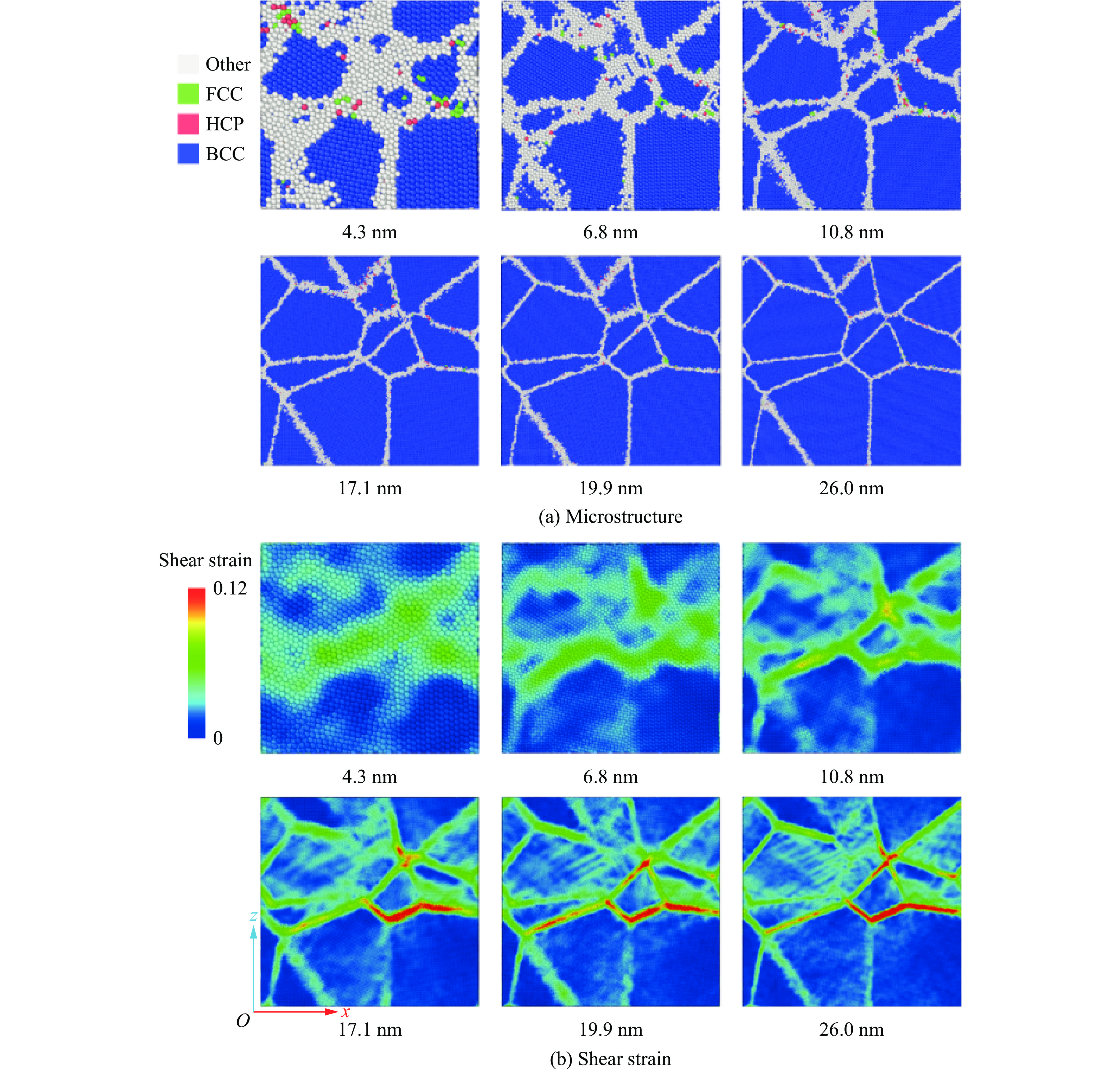

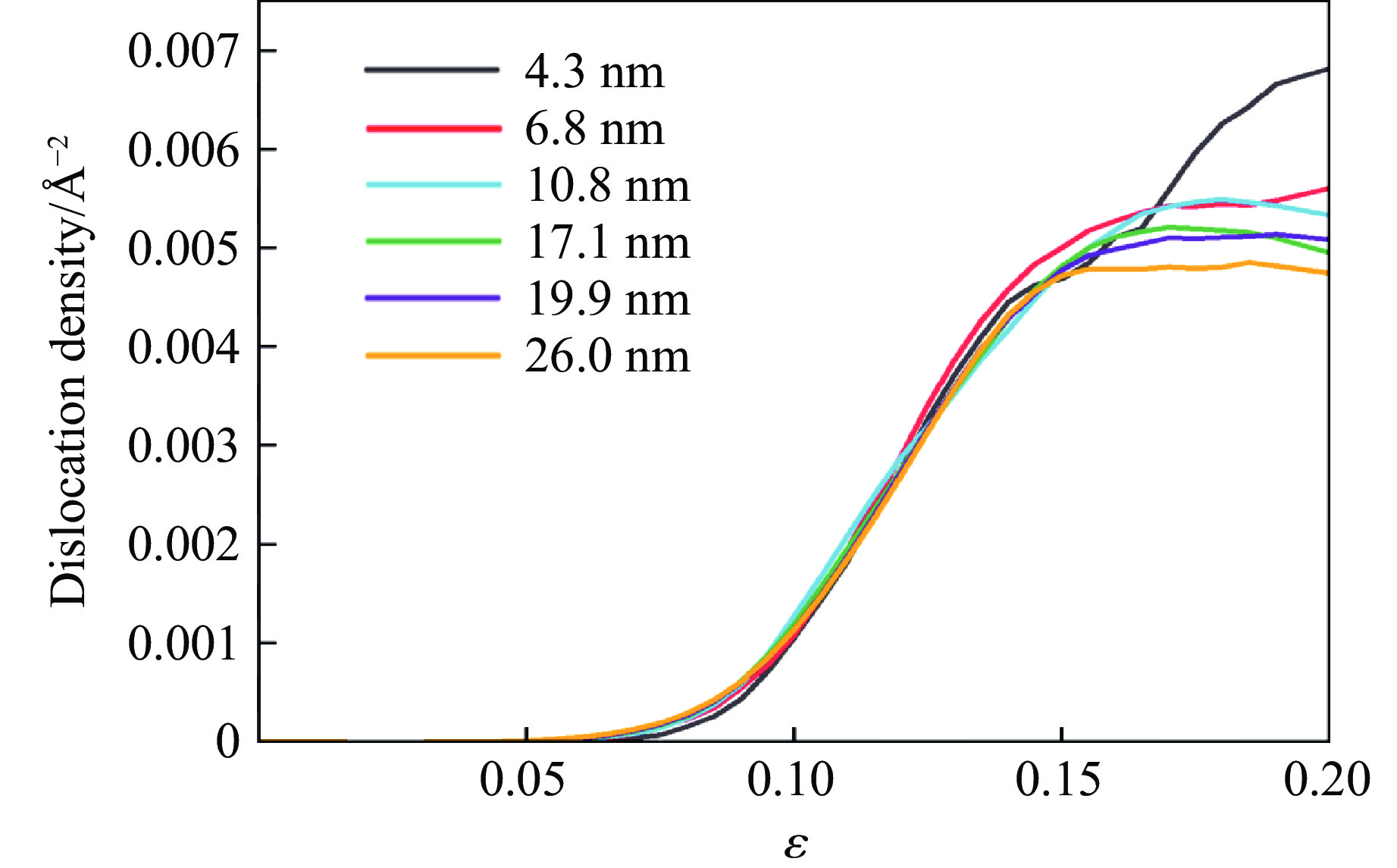

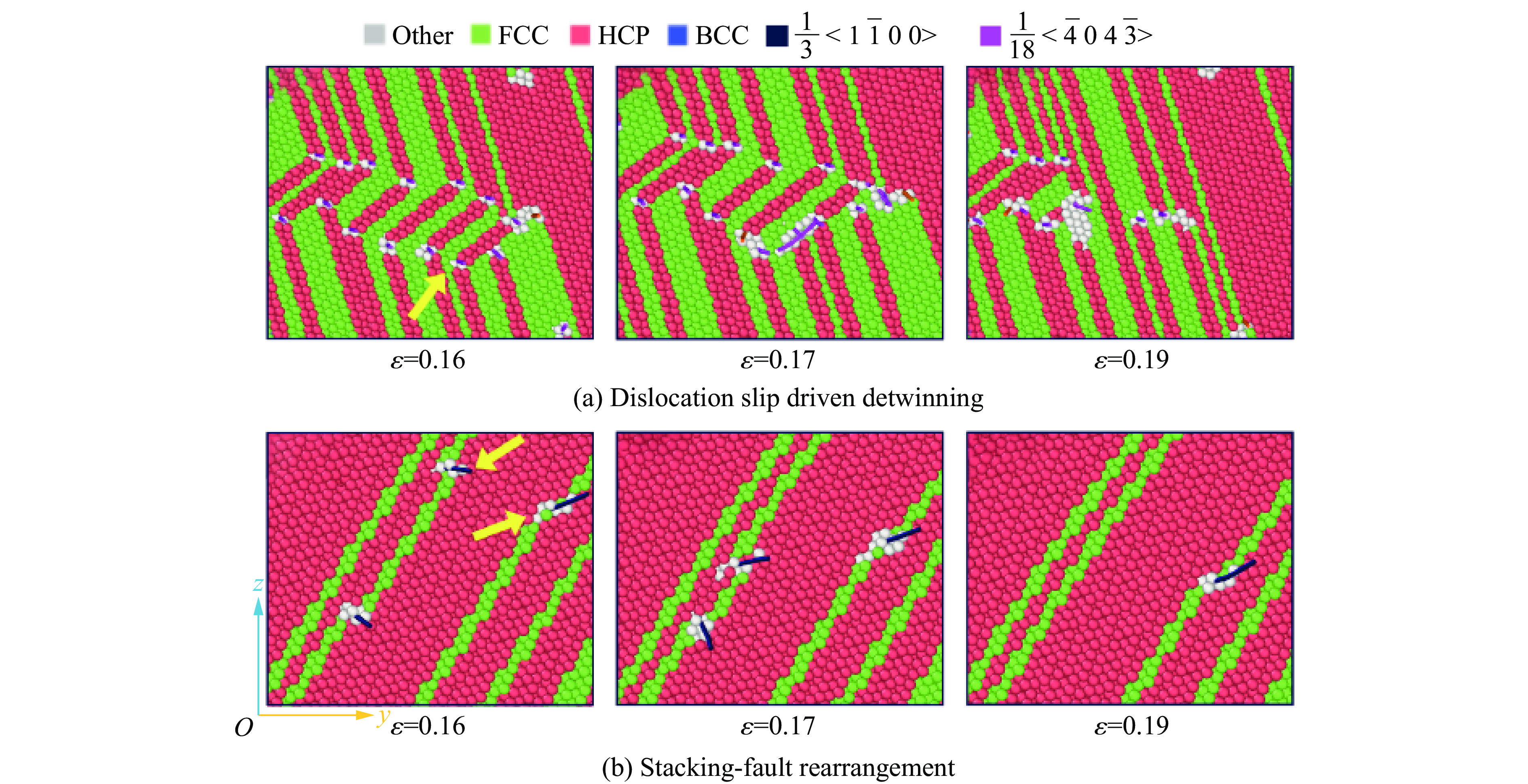
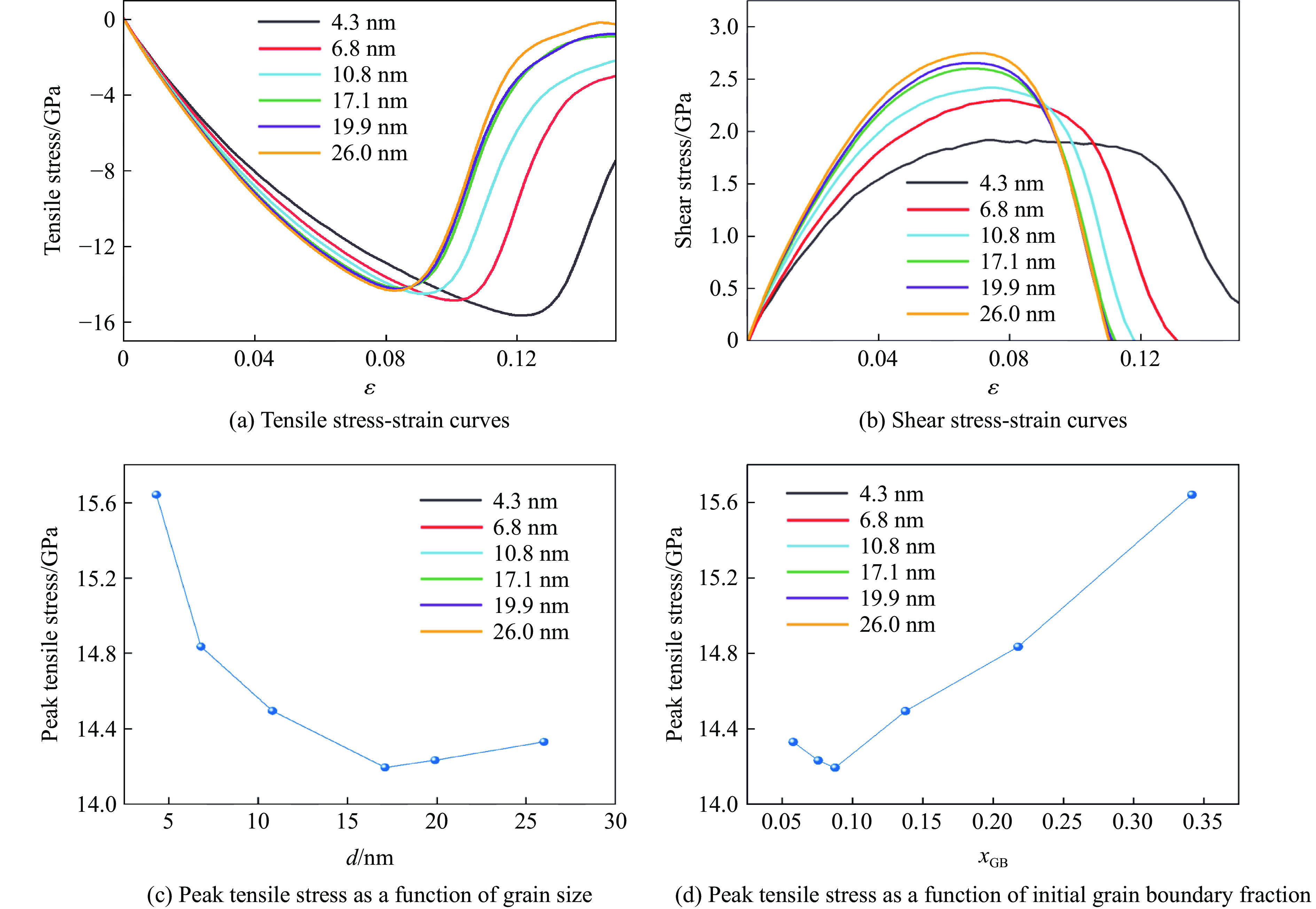

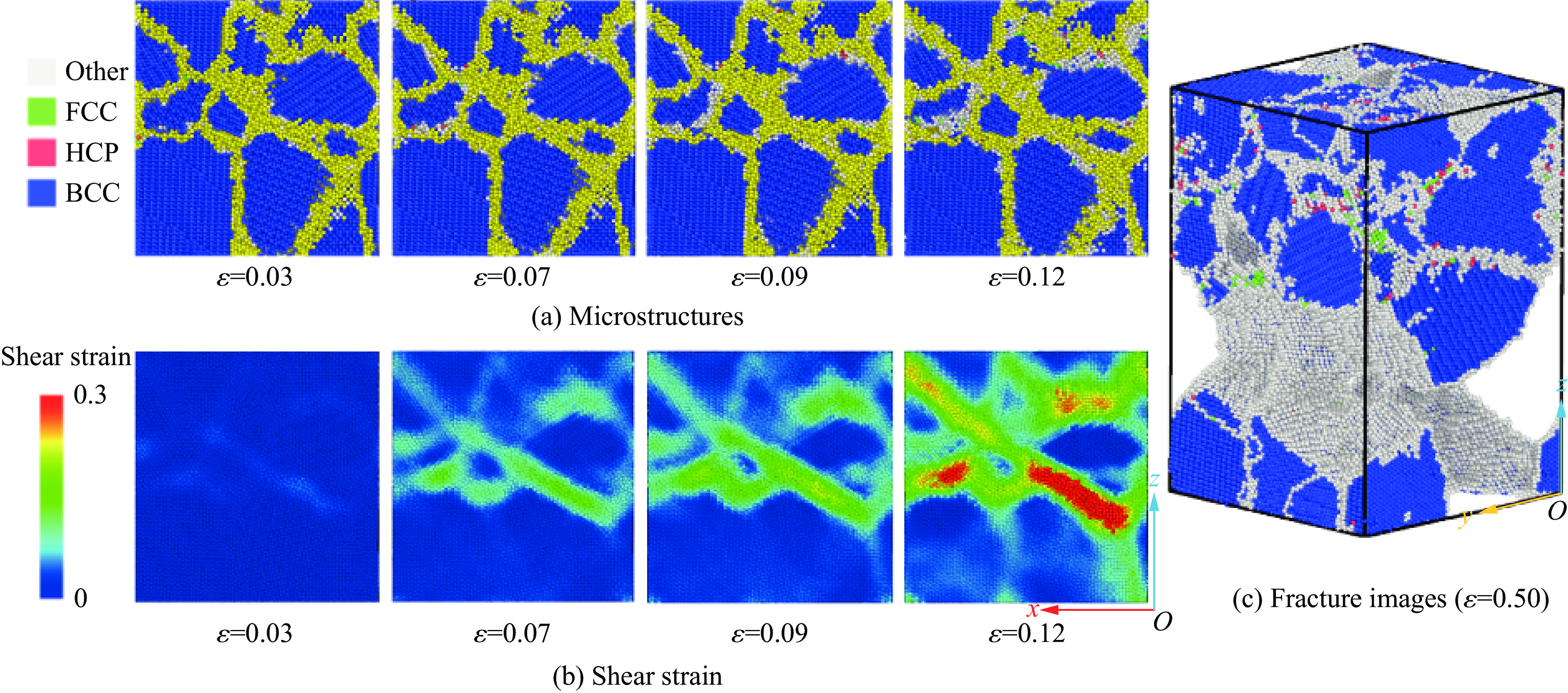
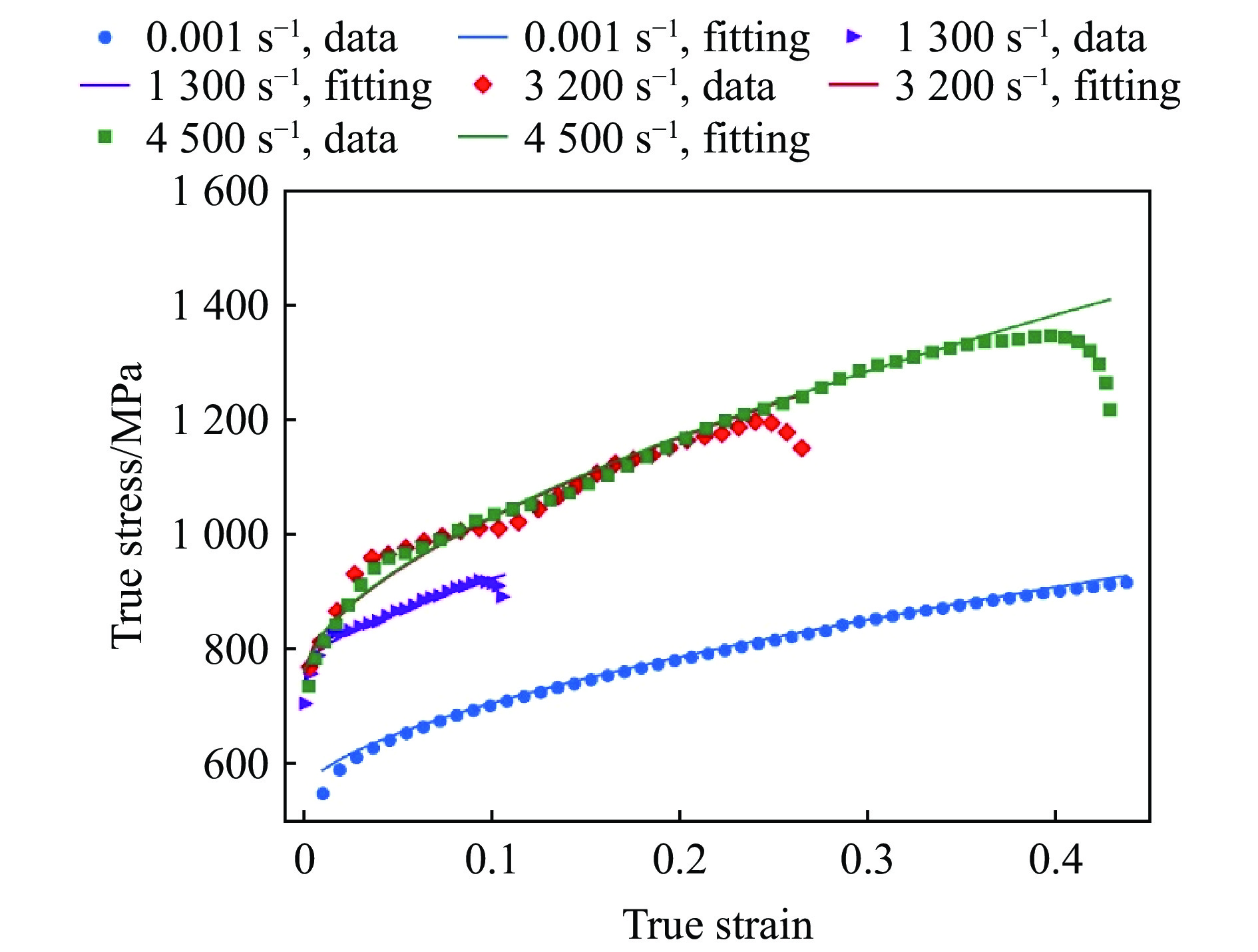
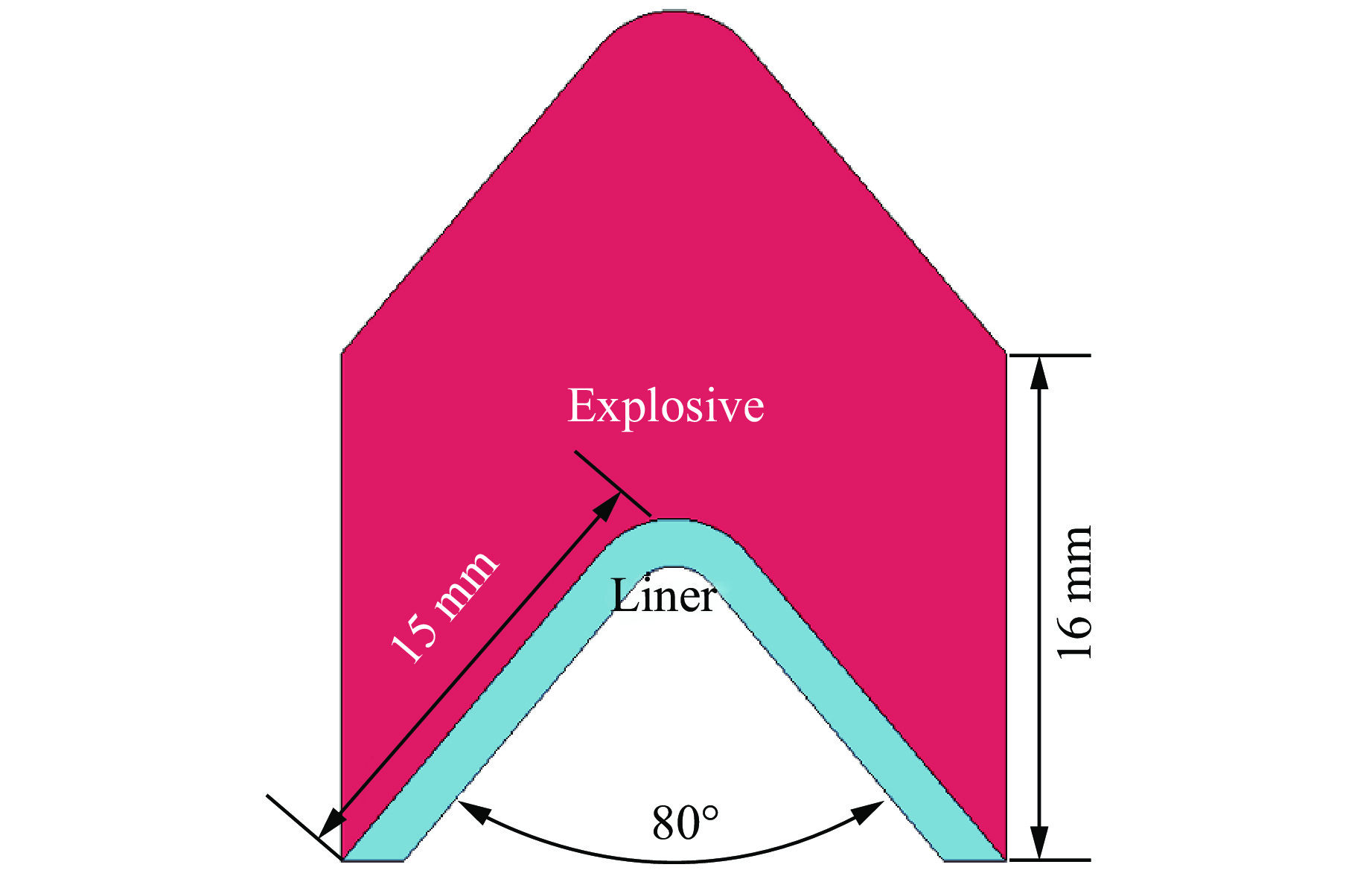
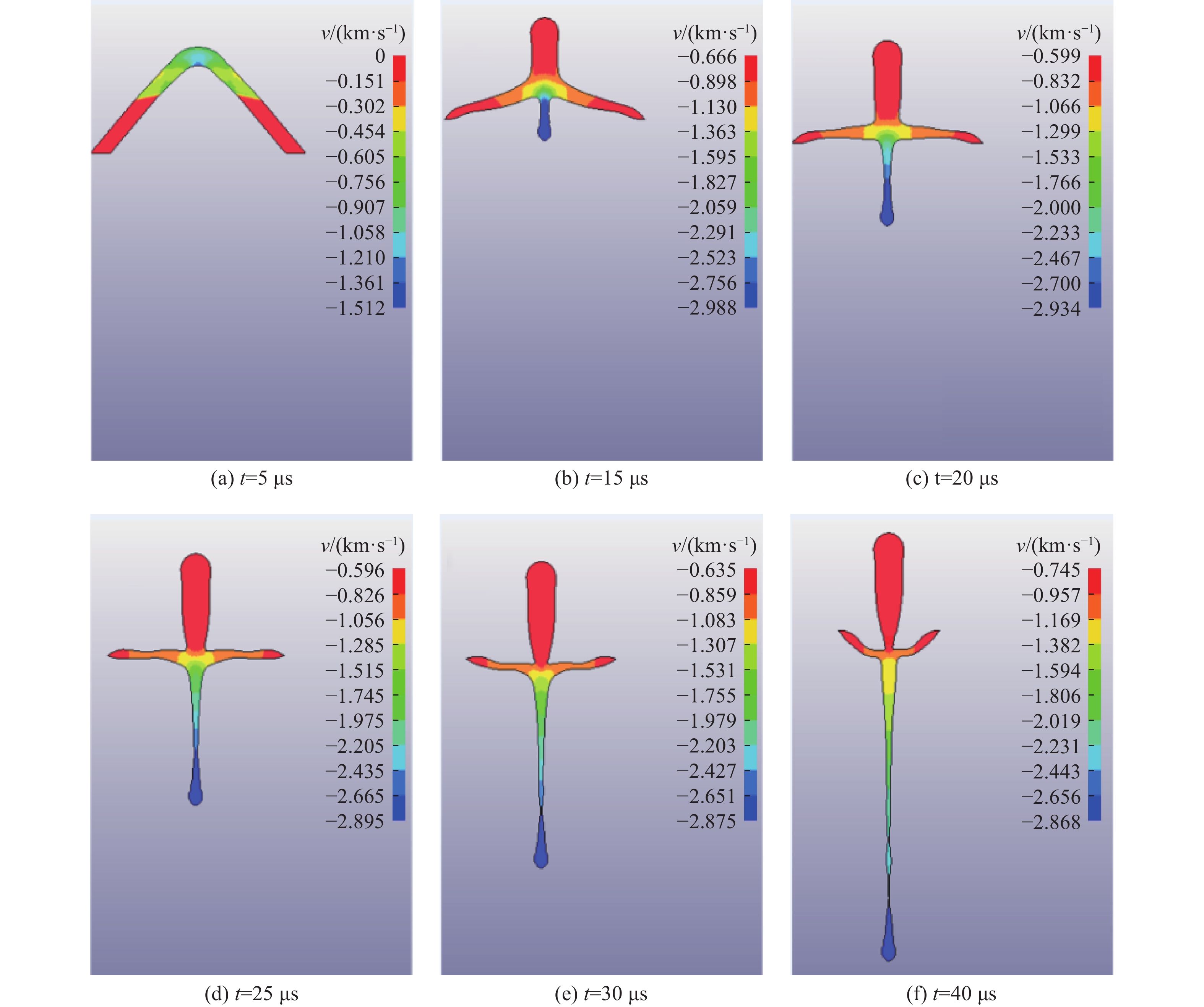
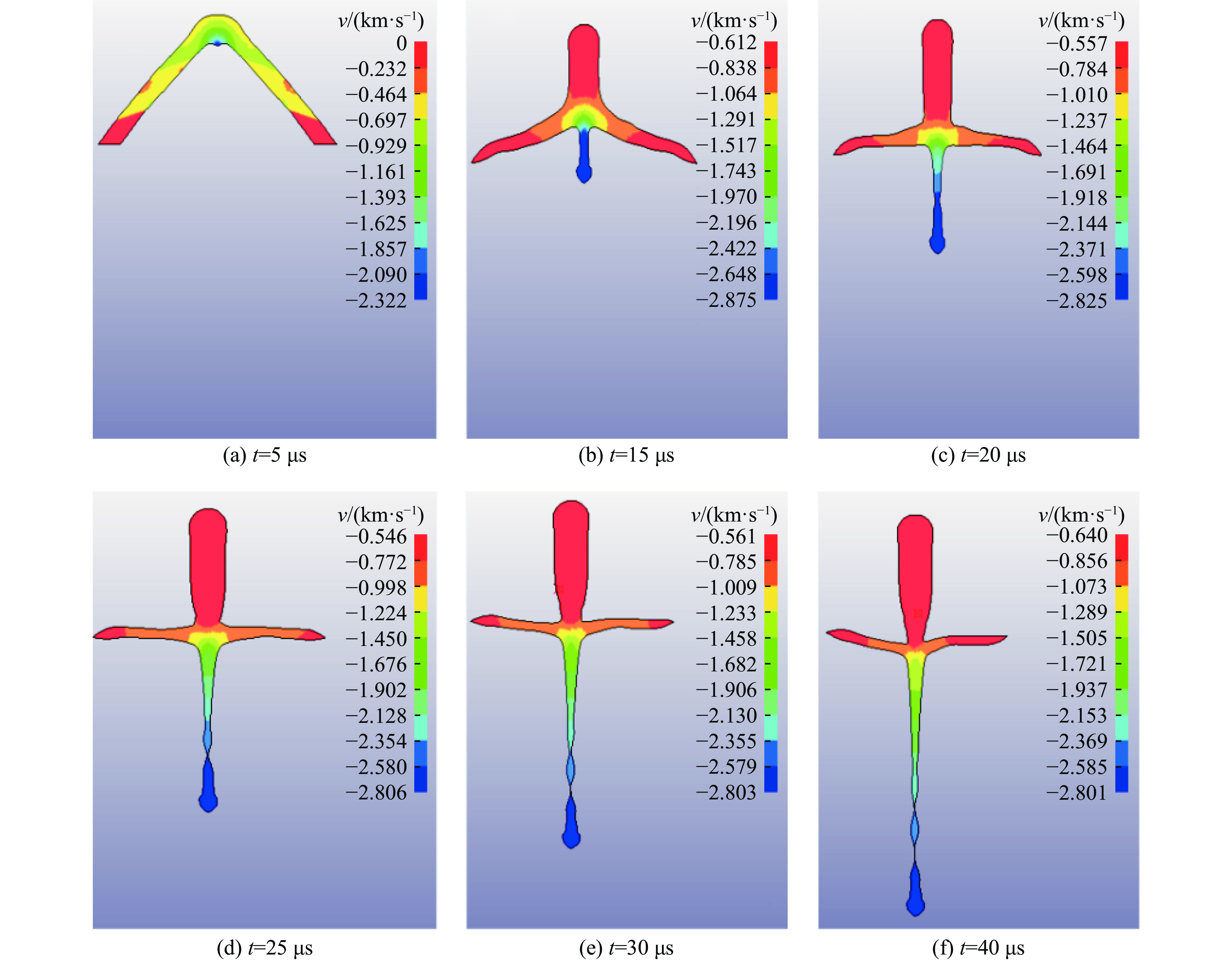
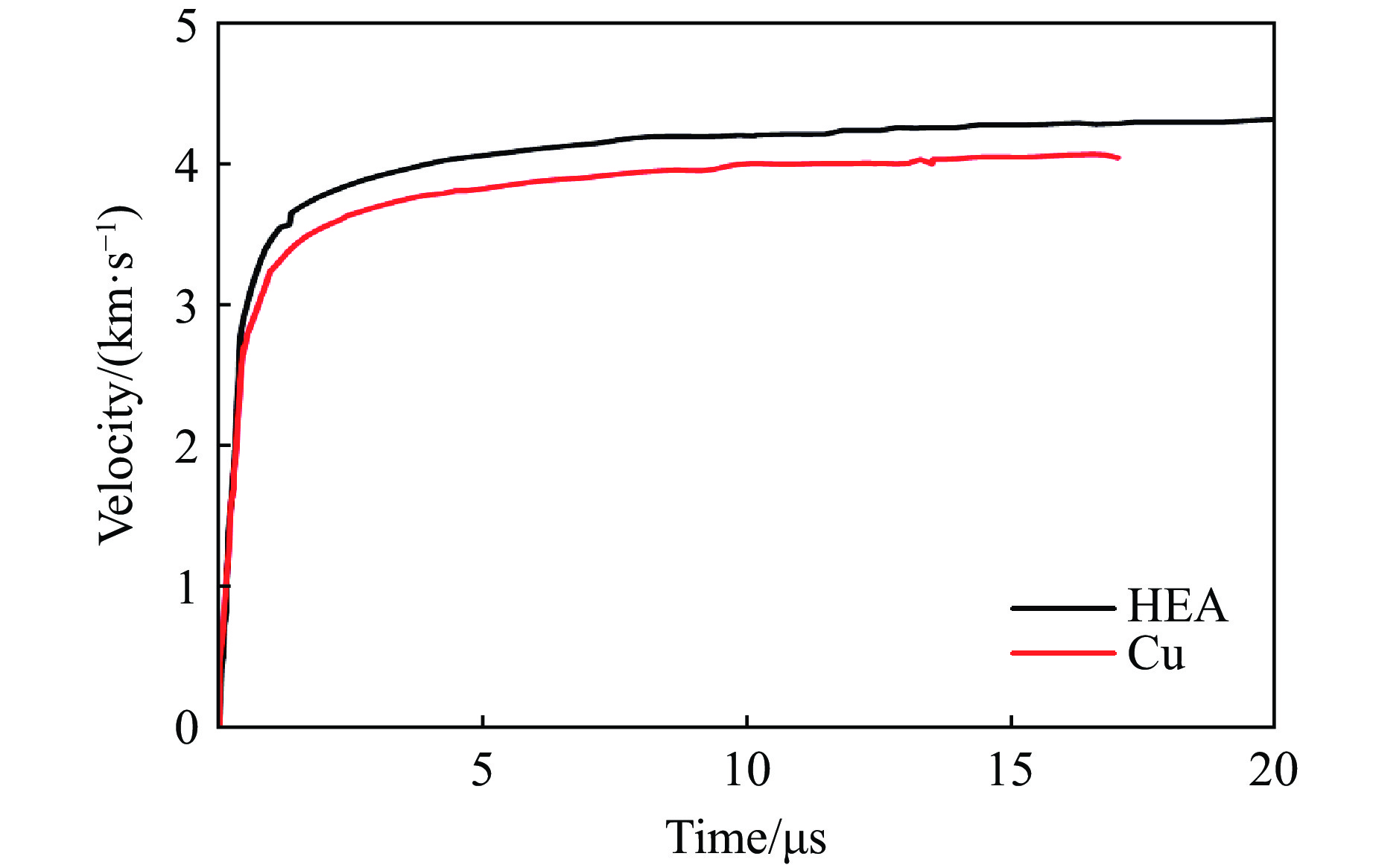
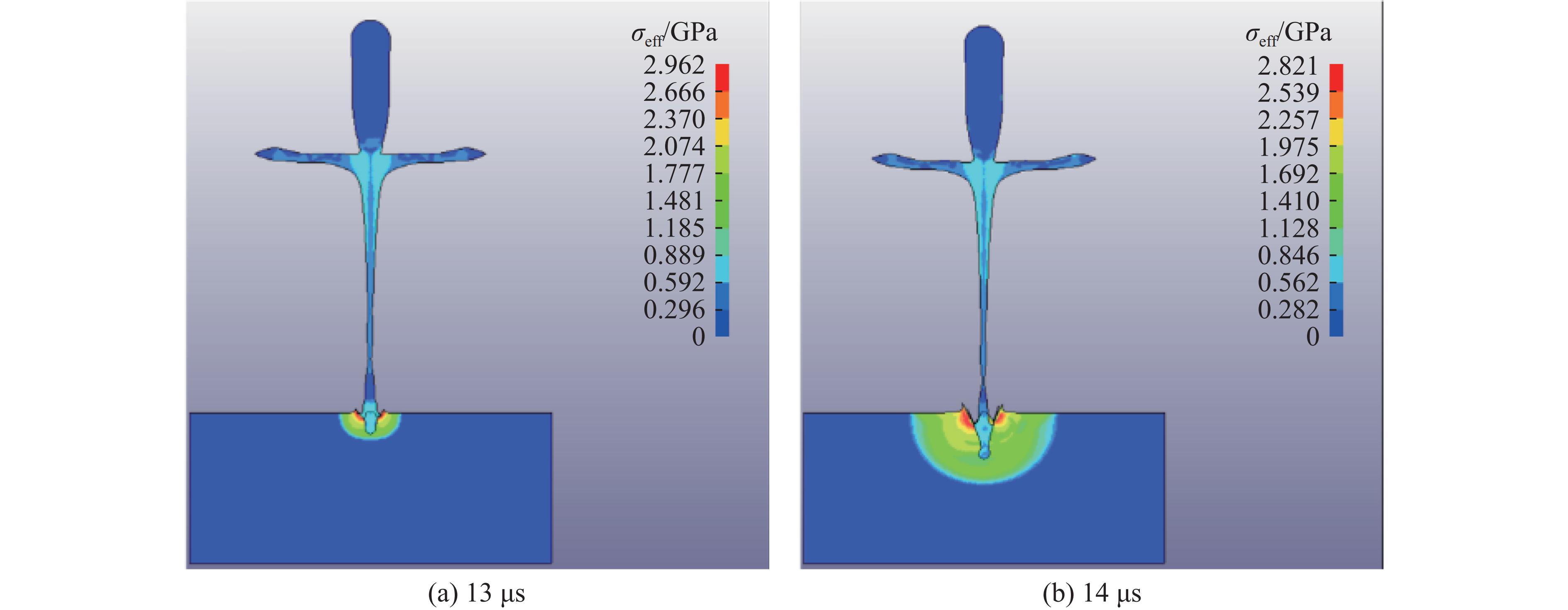
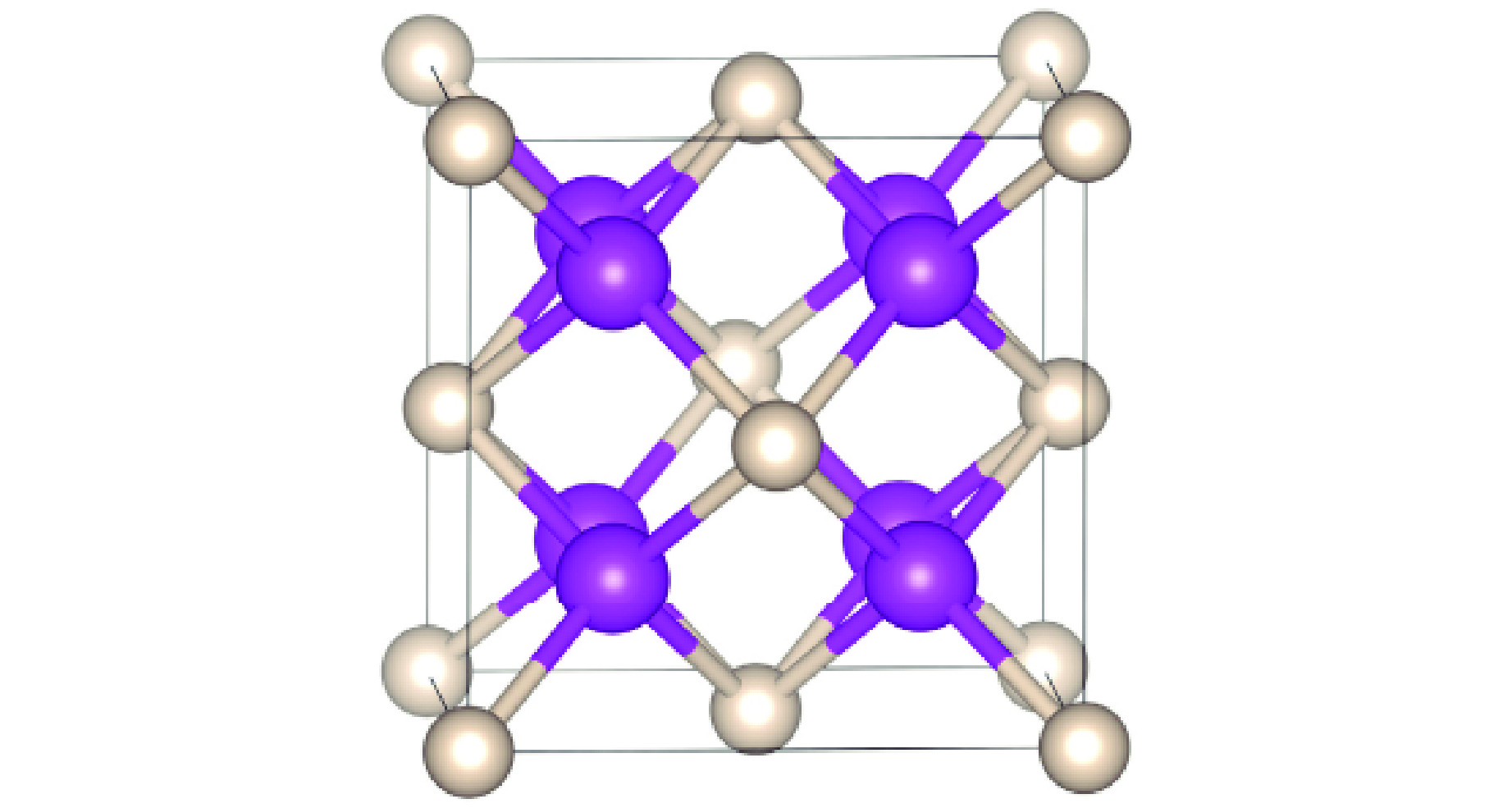
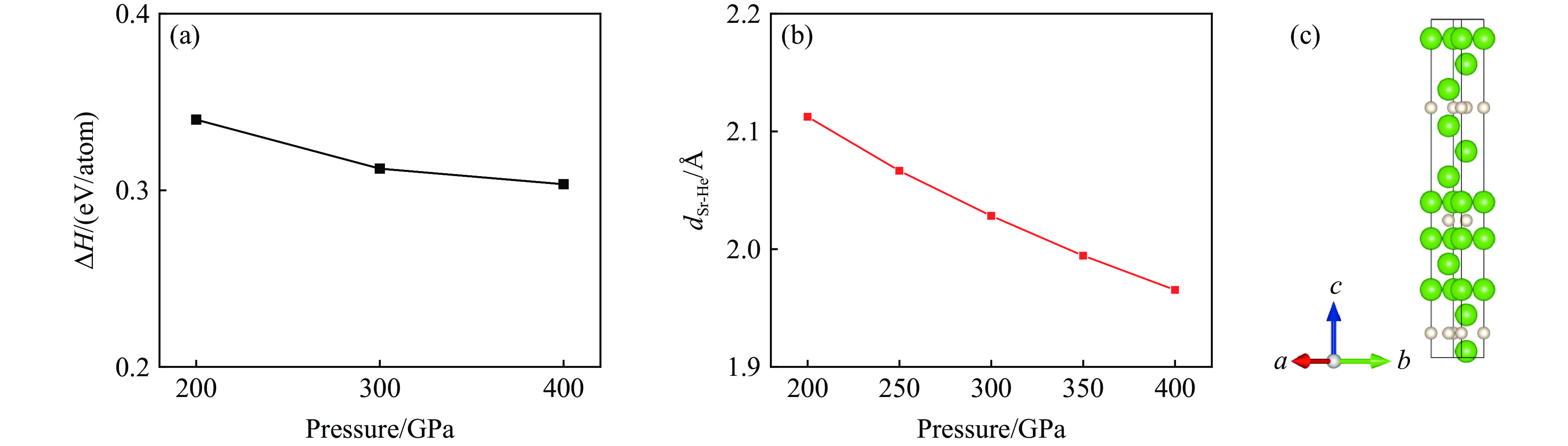
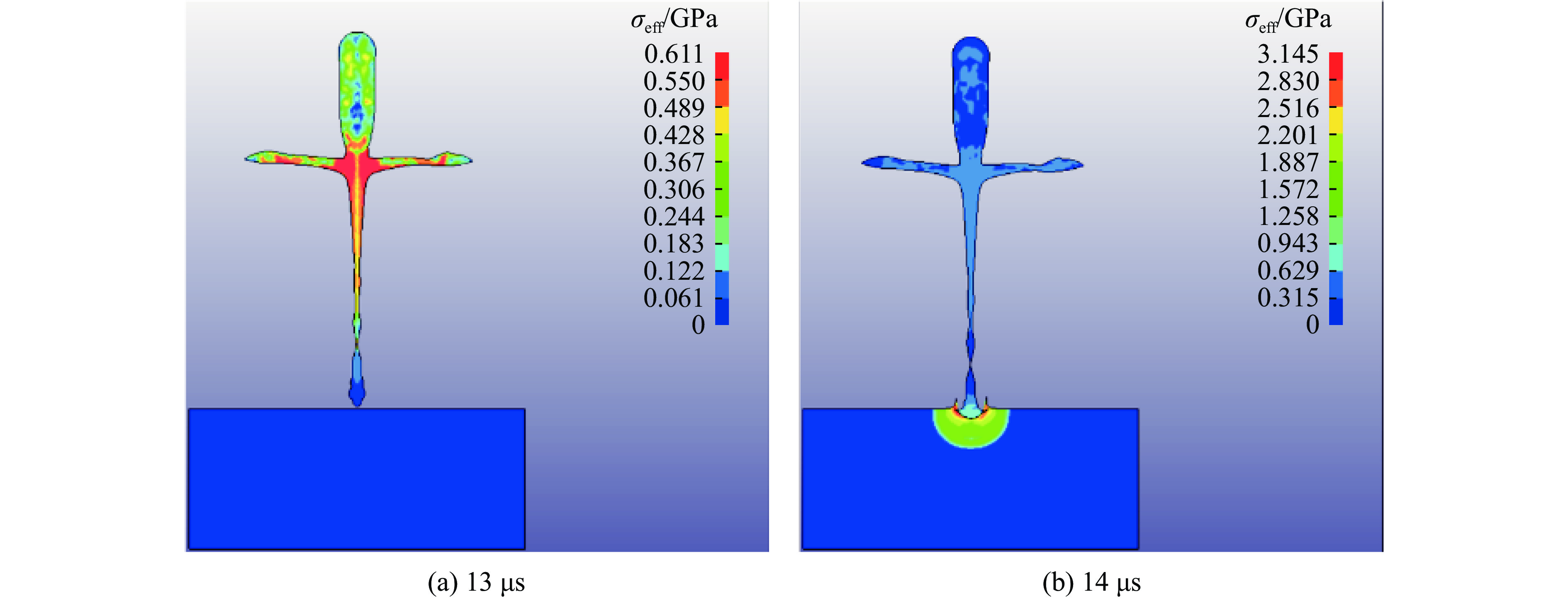 PDF(82)
PDF(82)



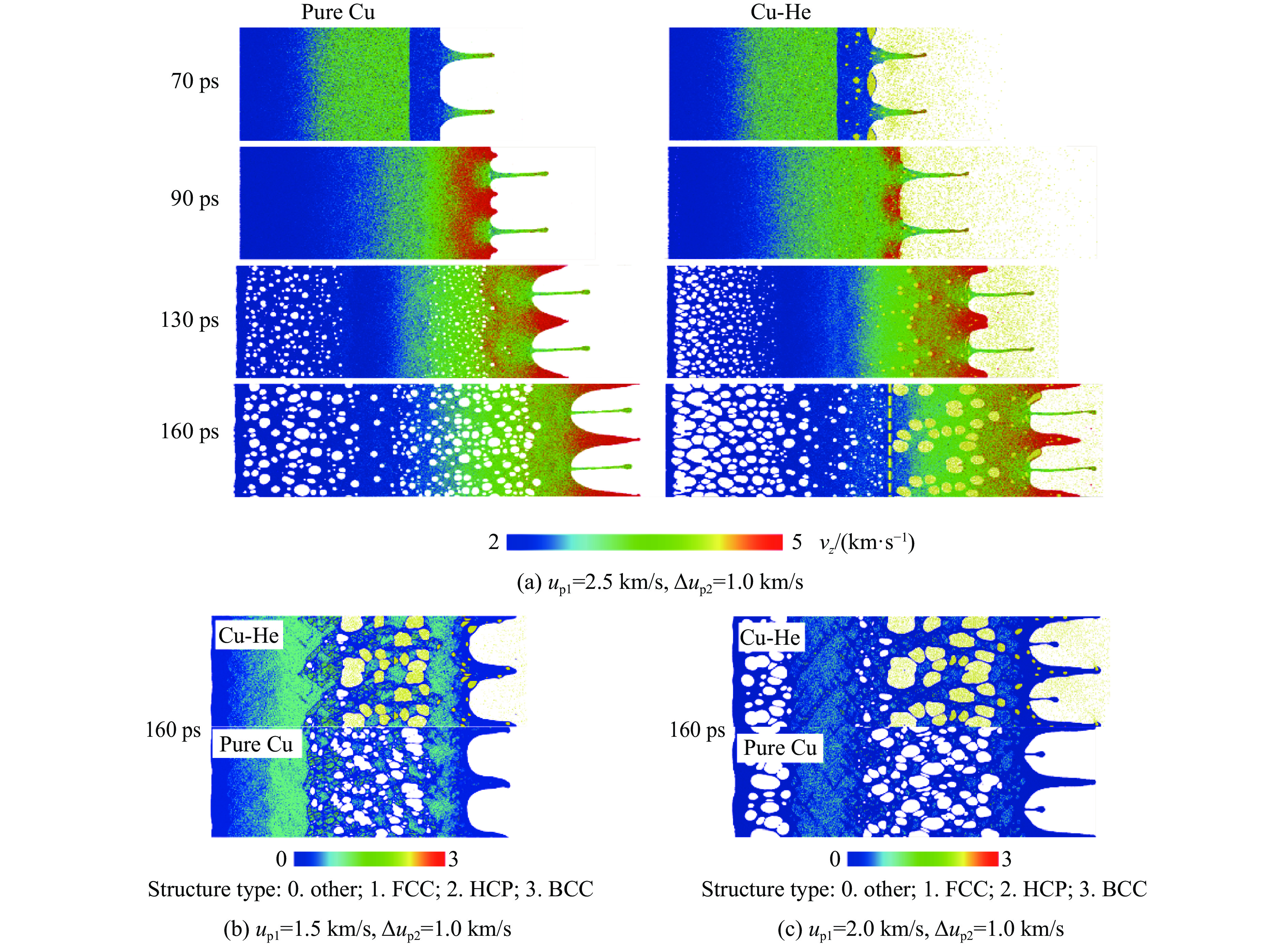
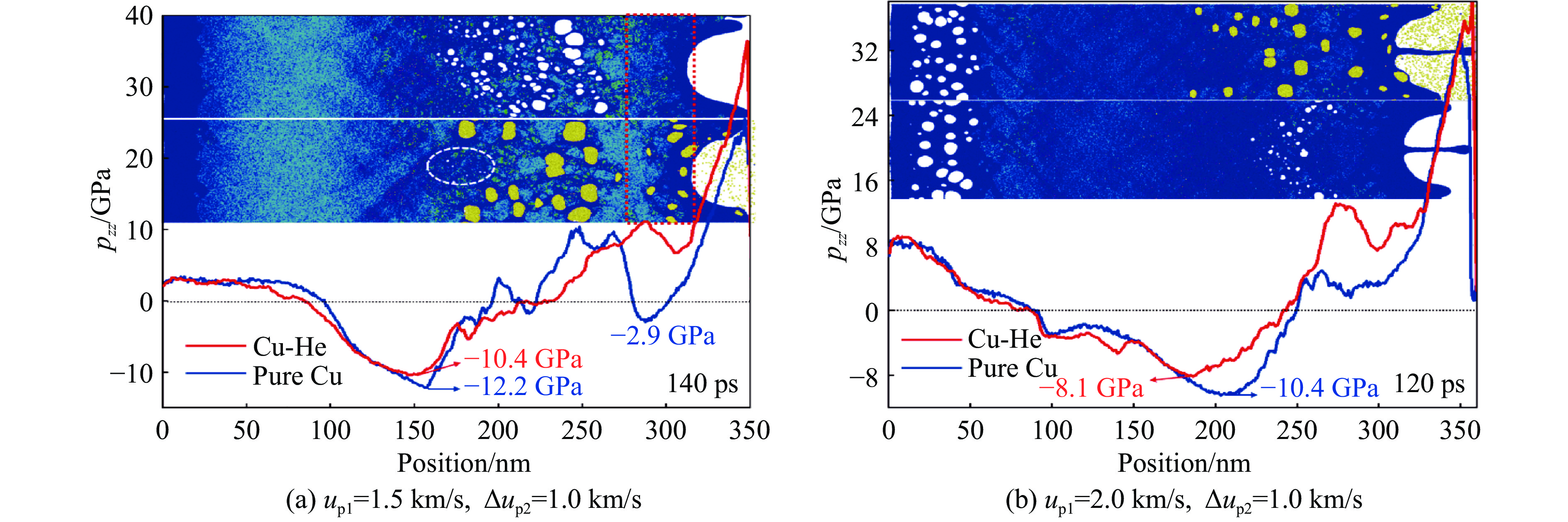

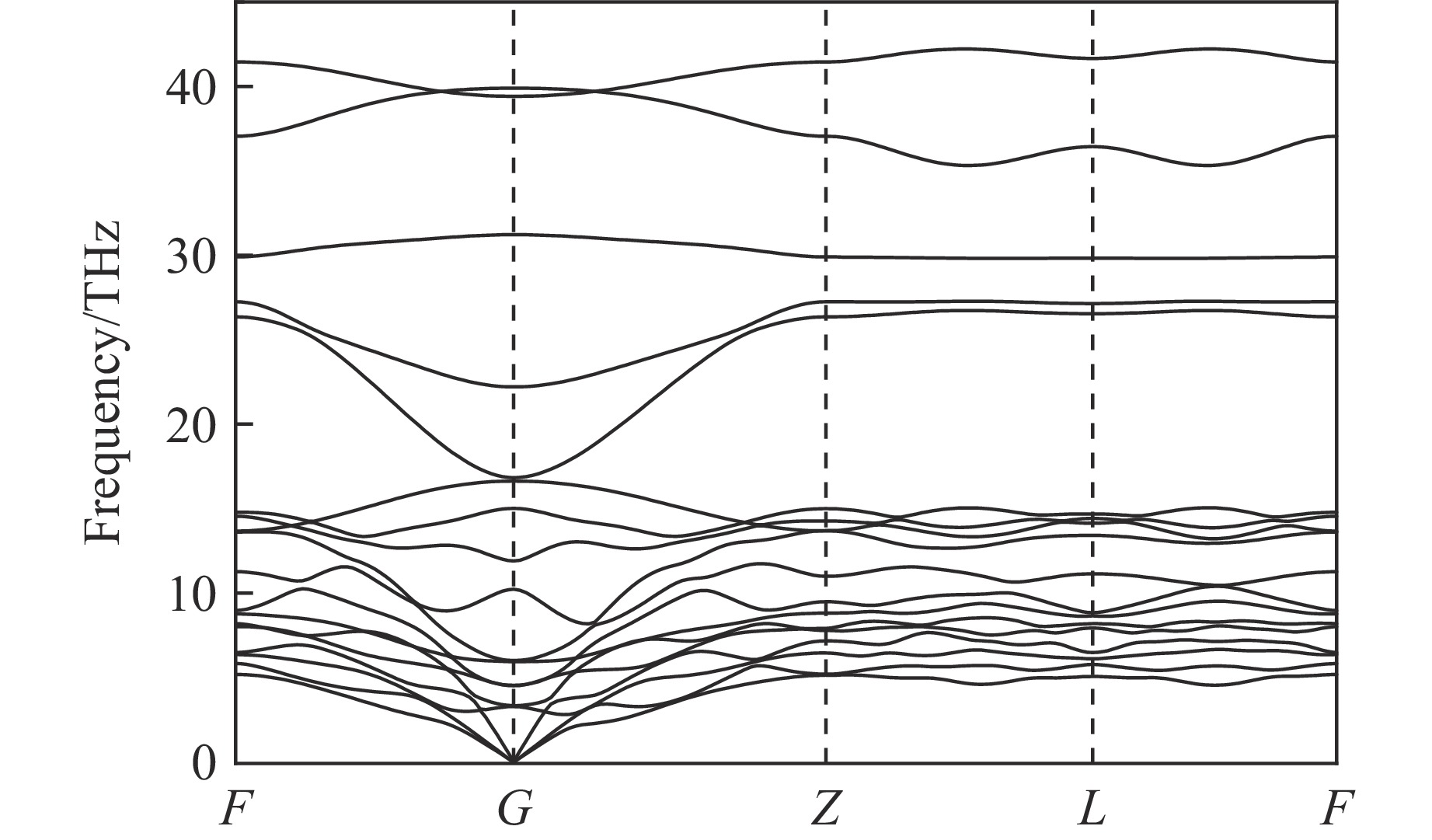
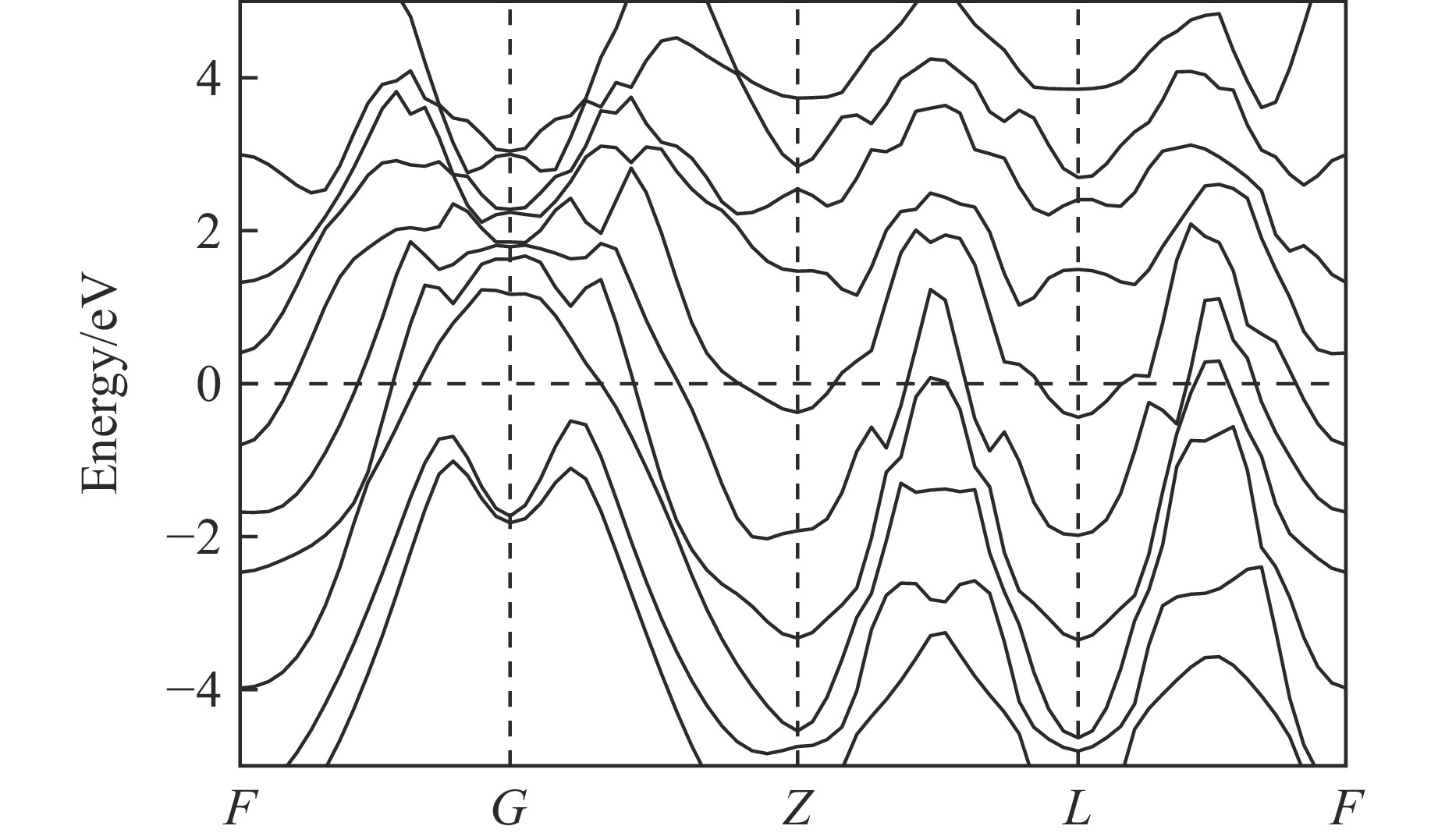
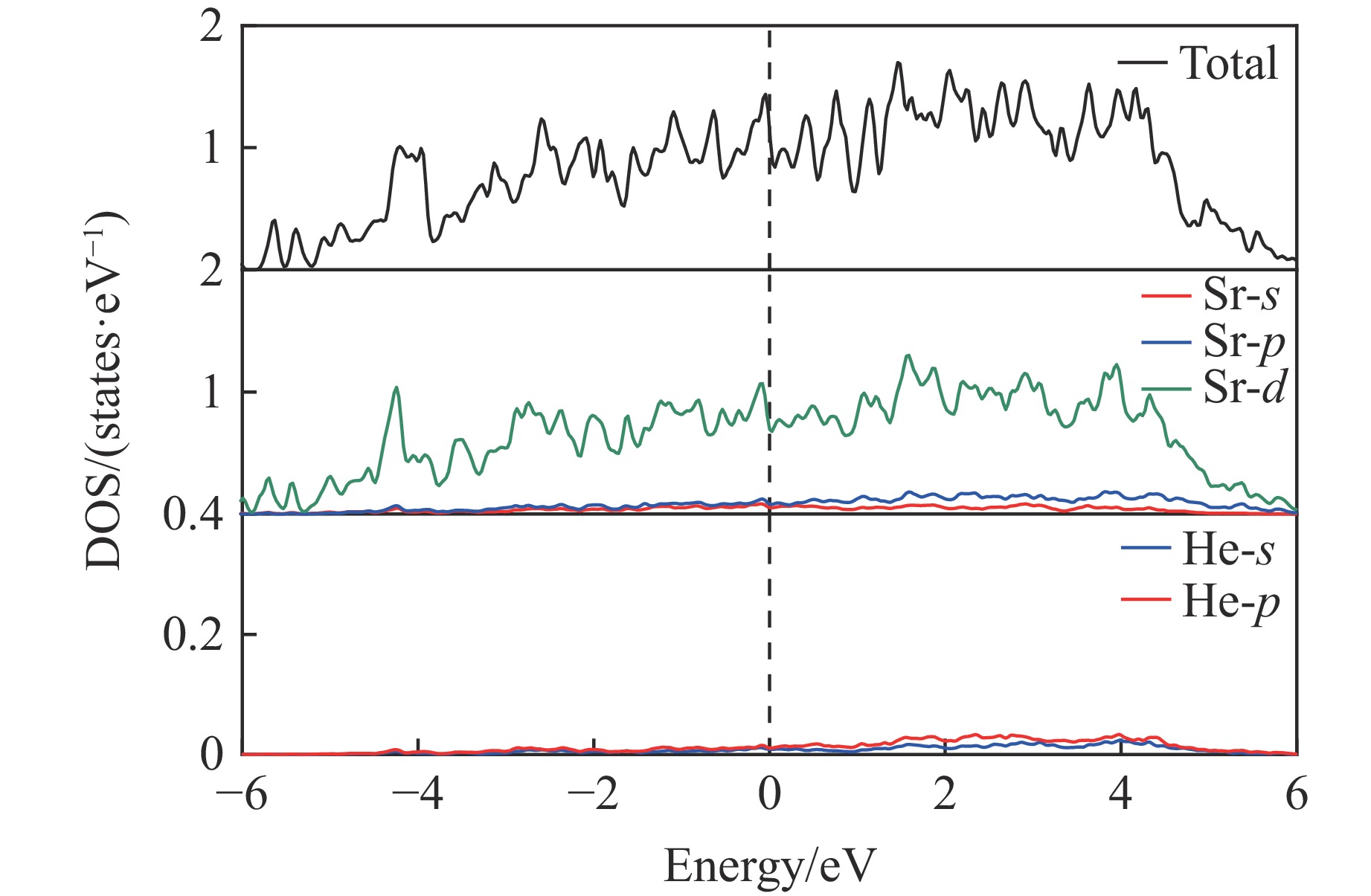
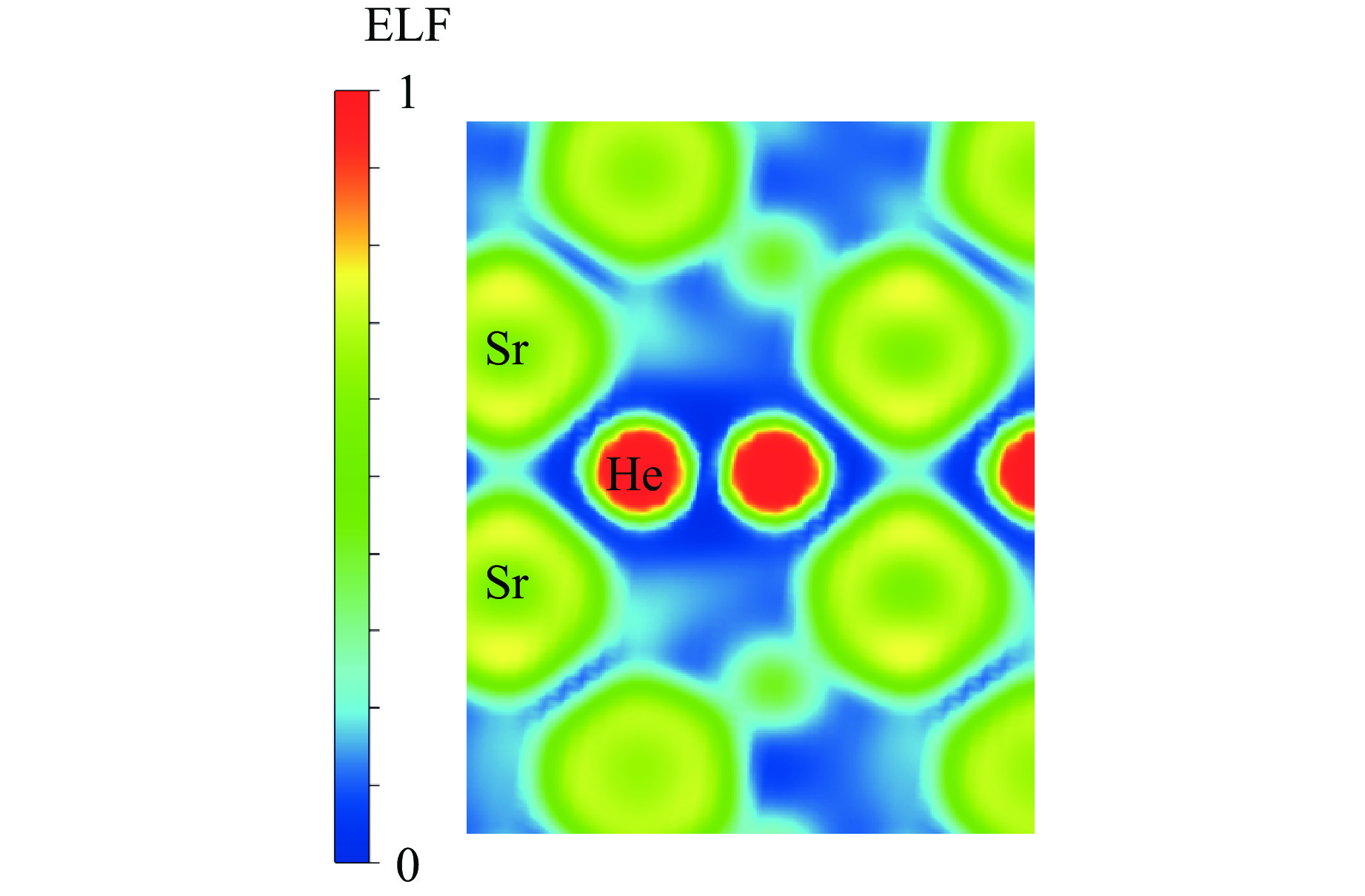
As lightweight and high-strength functional-structural integrated materials, cellular structural materials are widely applied in aerospace, automotive manufacturing, and biomedical fields. However, traditional single-configuration cellular materials (e.g., honeycomb structures and point-lattice lattices) gradually exhibit performance limitations under complex conditions such as impact shock waves, multi-directional impacts, or nonlinear deformations. Against this backdrop, heterogeneous cellular structure material (HCSM) have emerged as a research hot pot in impact protection. This paper systematically reviews recent design strategies and impact resistance performance of HCSM. HCSMs are primarily categorized into two types: topological configuration heterogeneity (including complementary and enhanced fusion) and material heterogeneity (e.g., filling with foam materials and shear-thickening materials). Through innovative “functional fusion” approaches, they overcome the performance bottlenecks of single-configuration cellular materials. The study further elucidates the synergistic reinforcement effects and deformation mechanisms of HCSM under impact loads, while analyzing their intrinsic mechanisms for improving energy absorption efficiency, stiffness, and stability. Despite significant progress in HCSM research, challenges remain in connectivity optimization, additive manufacturing process compatibility, complex condition validation, and multifunctional integration. Going forward, the integration of artificial intelligence and machine learning technologies holds promise for achieving end-to-end optimization of HCSMs from design to manufacturing, thereby providing new directions for developing next-generation high-performance impact-resistant structural materials.






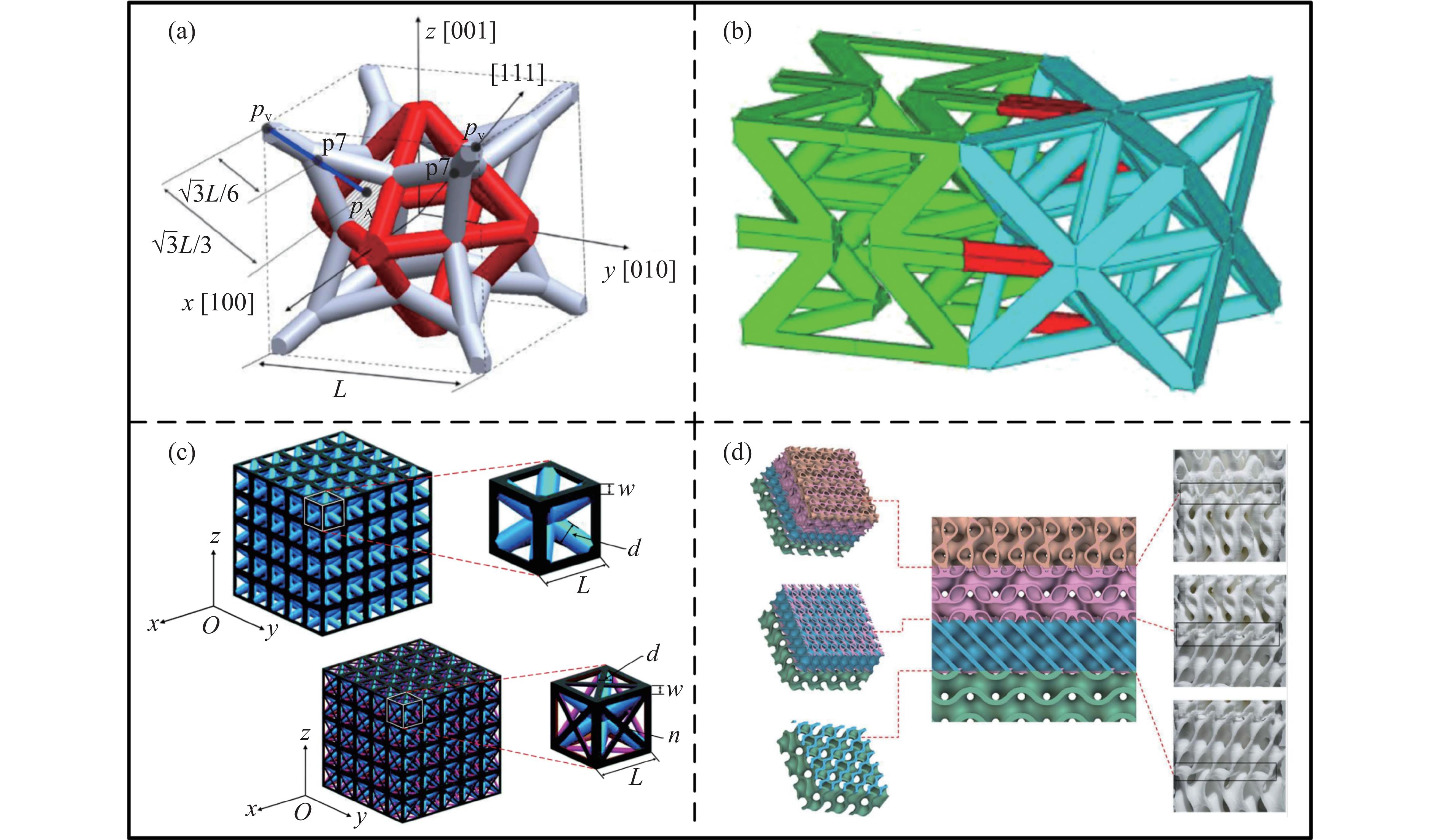
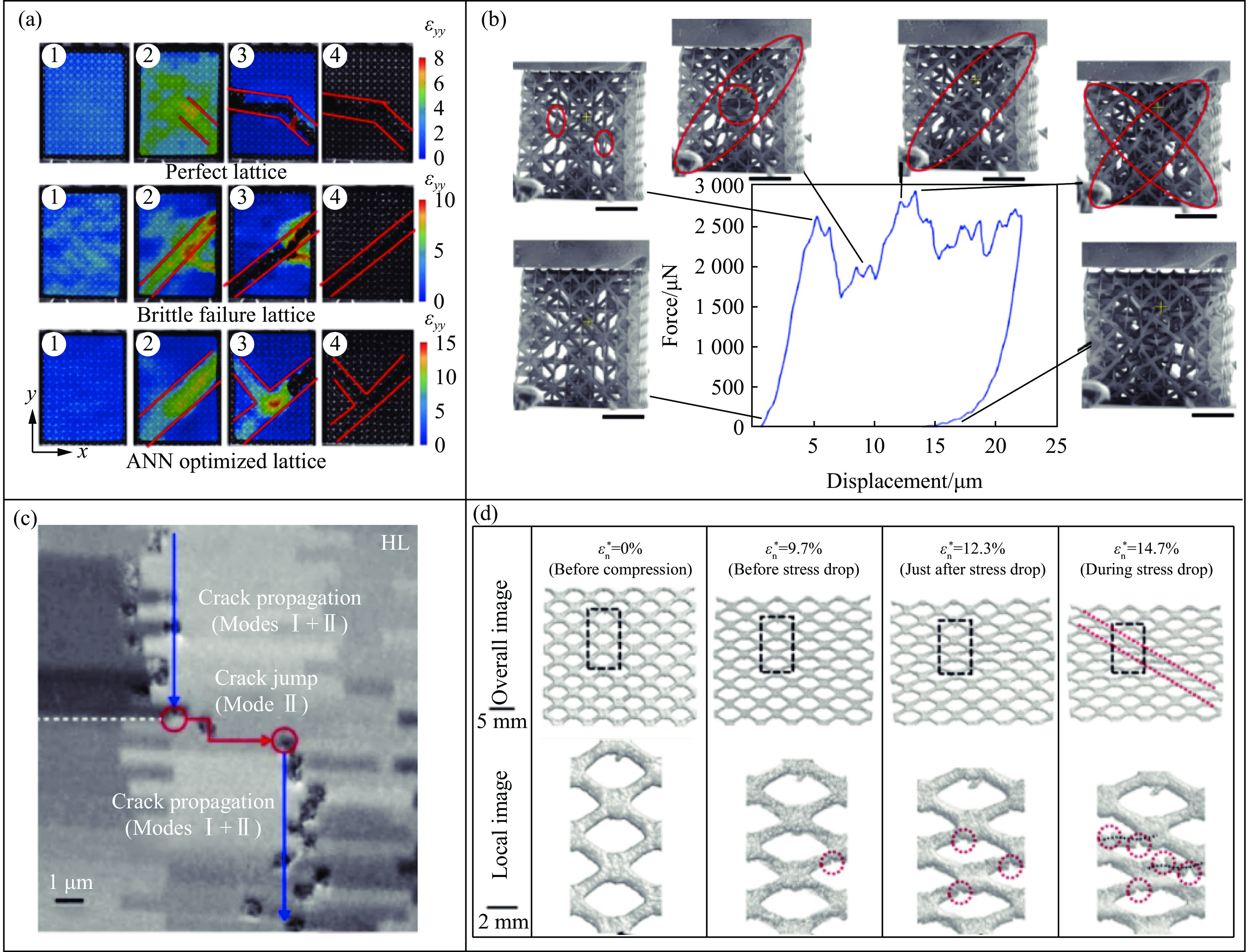
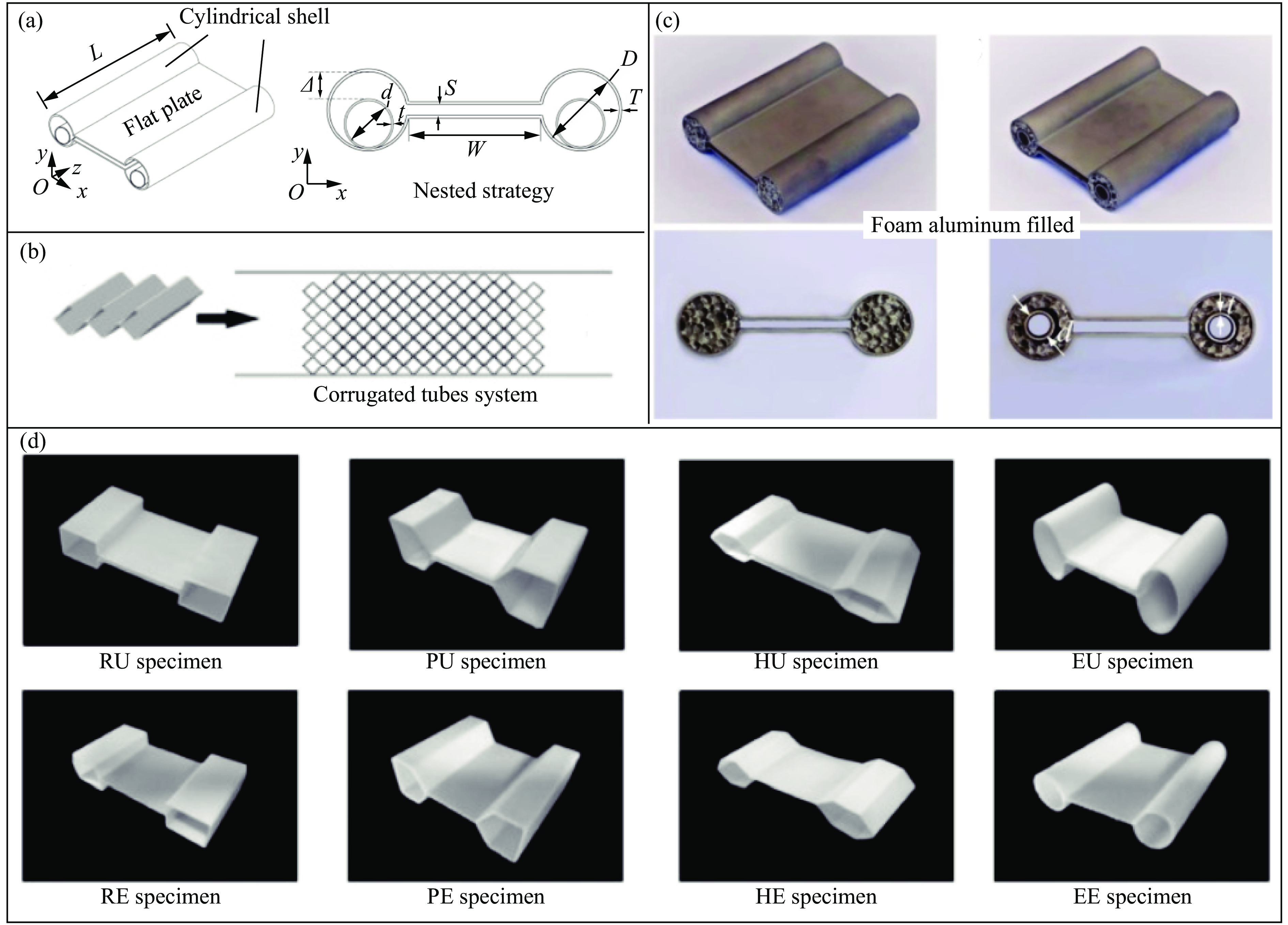
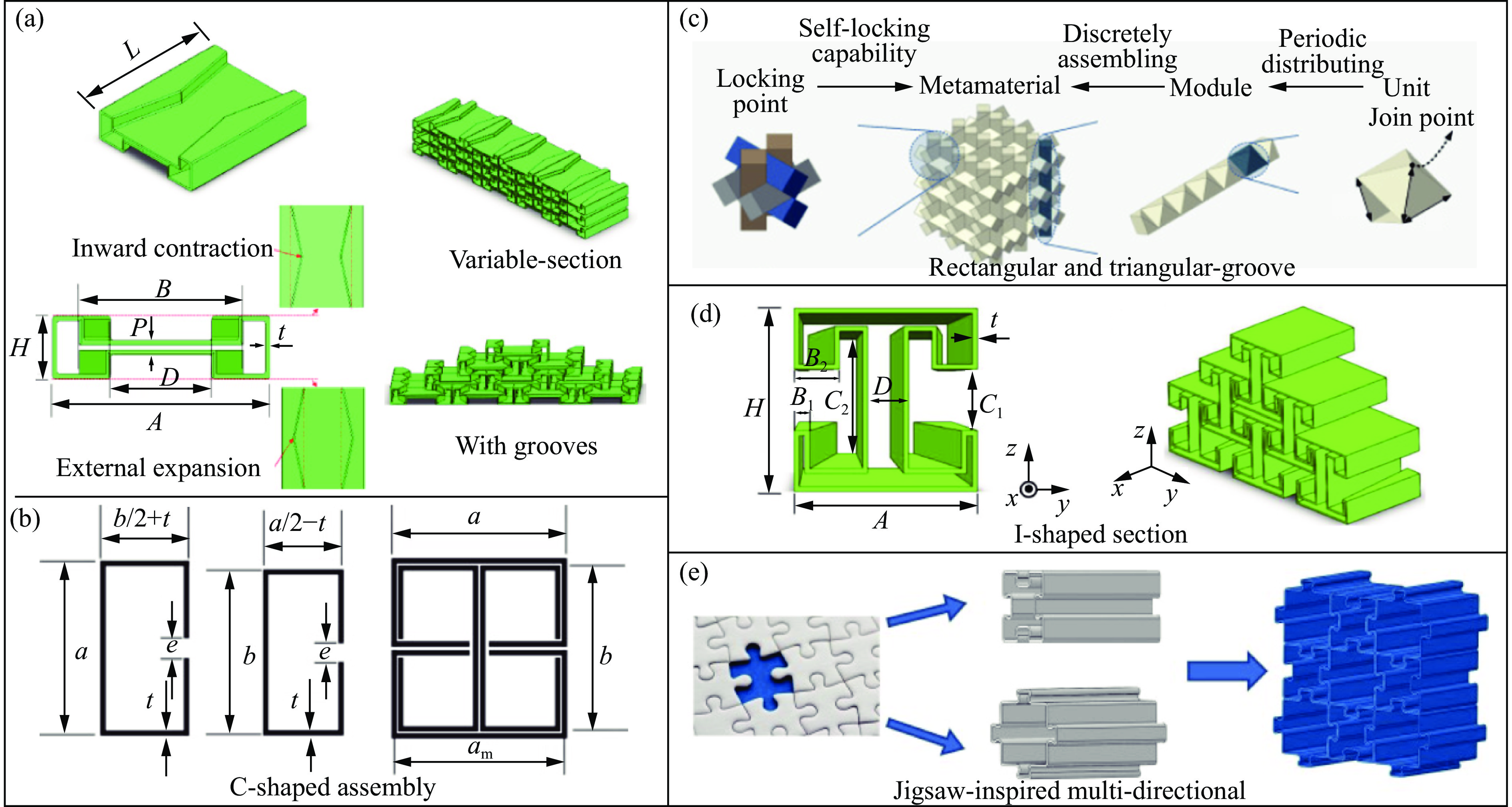
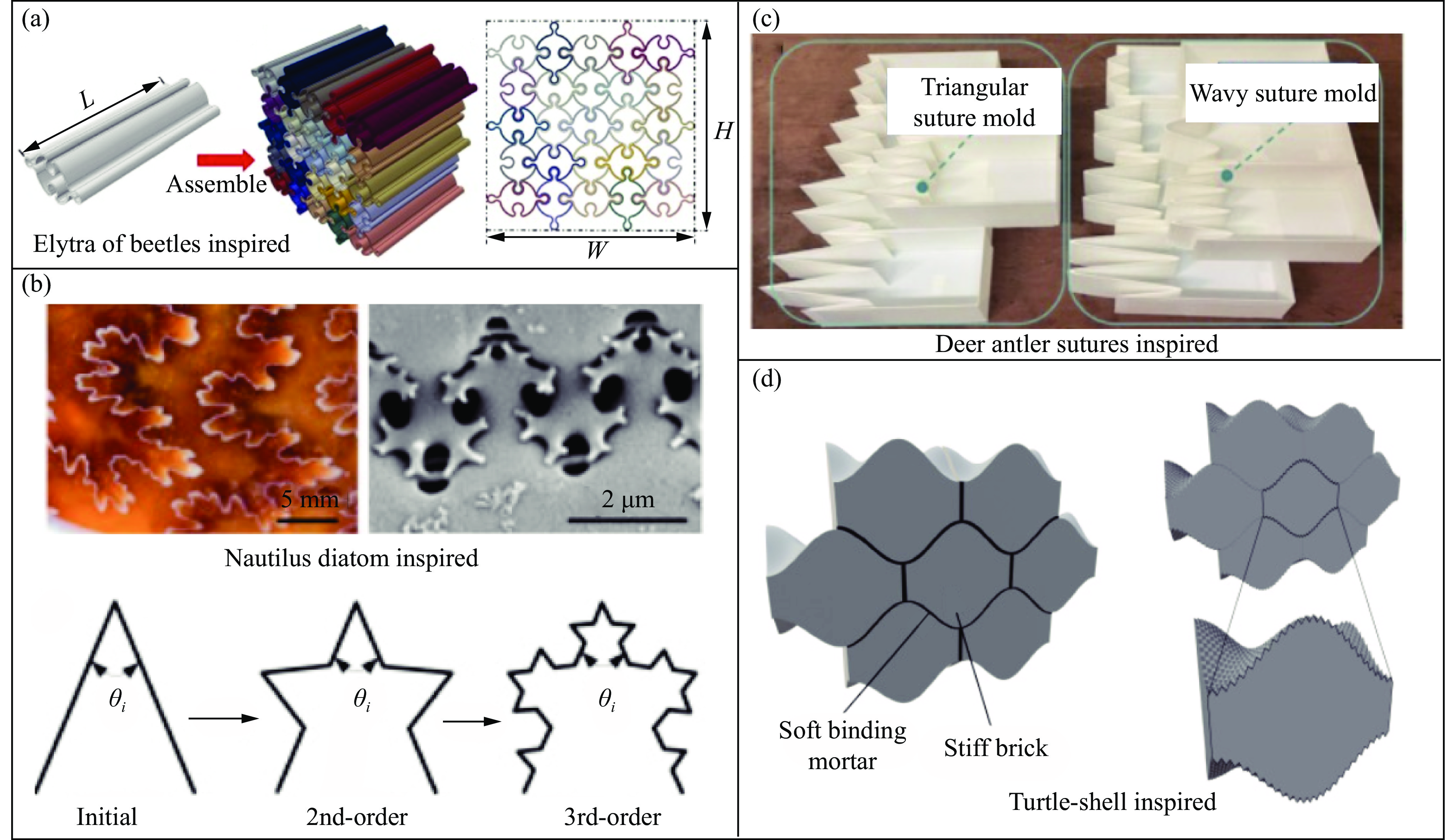
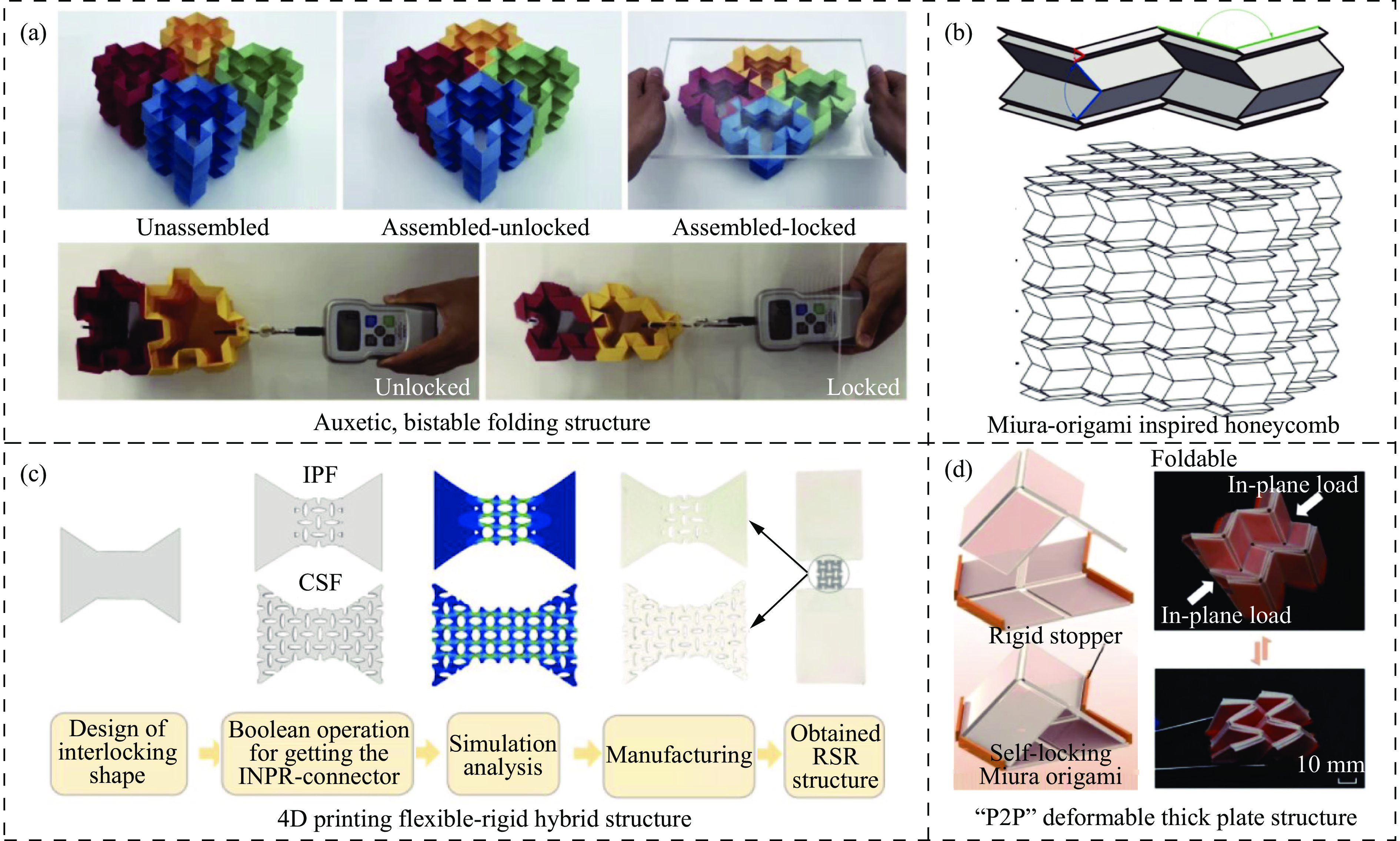
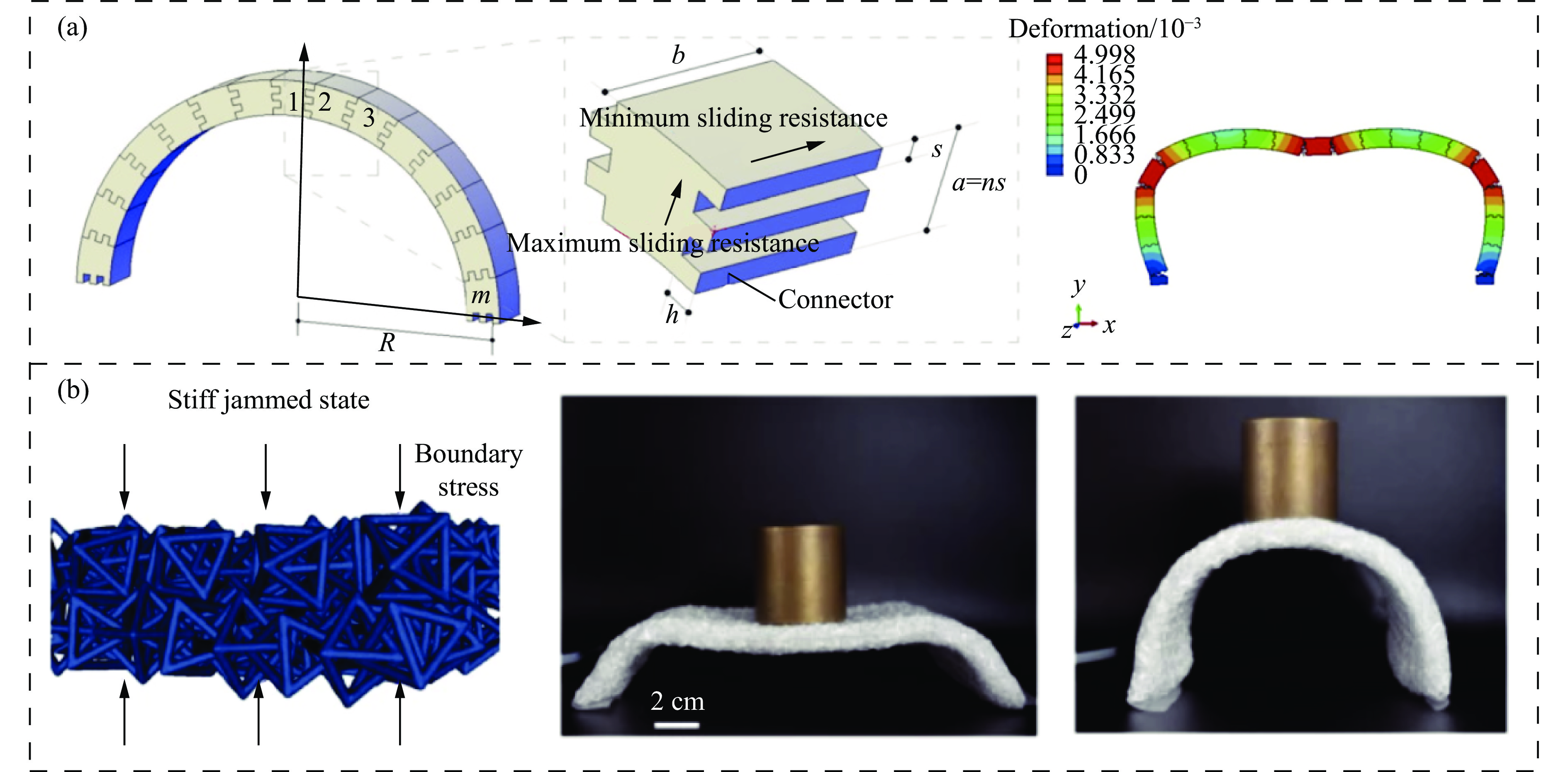
The self-locking structure achieves interlocking property through ingenious design of the connection mode between cells, which enables the cells to lock with each other without the need for any additional constraints. The self-locking structure possesses significant advantages, such as light weight, portability, rapid assembly, and disassembly. Therefore, this structure is widely applied in various fields, such as shock resistance and explosion prevention. Self-locking structures exist in many structures in nature. The design concepts of self-locking structures are introduced from three aspects: the inspiration from biomimetic self-locking structures, the energy absorption mechanism of periodic structures, and failure of shear bands in periodic structures. The research progress of two-dimensional unidirectional self-locking structures, three-dimensional multi-directional self-locking structures and curved self-locking structures are then respectively introduced based on the classification of self-locking direction. Among them, the multi-directional self-locking structure withstands more complex loading conditions. Therefore, research progress of three representative multi-directional self-locking structures based on dumbbell-type, bone stitching, and origami design are further introduced. Finally, the research on the self-locking structure is summarized, and its future research prospects are discussed.
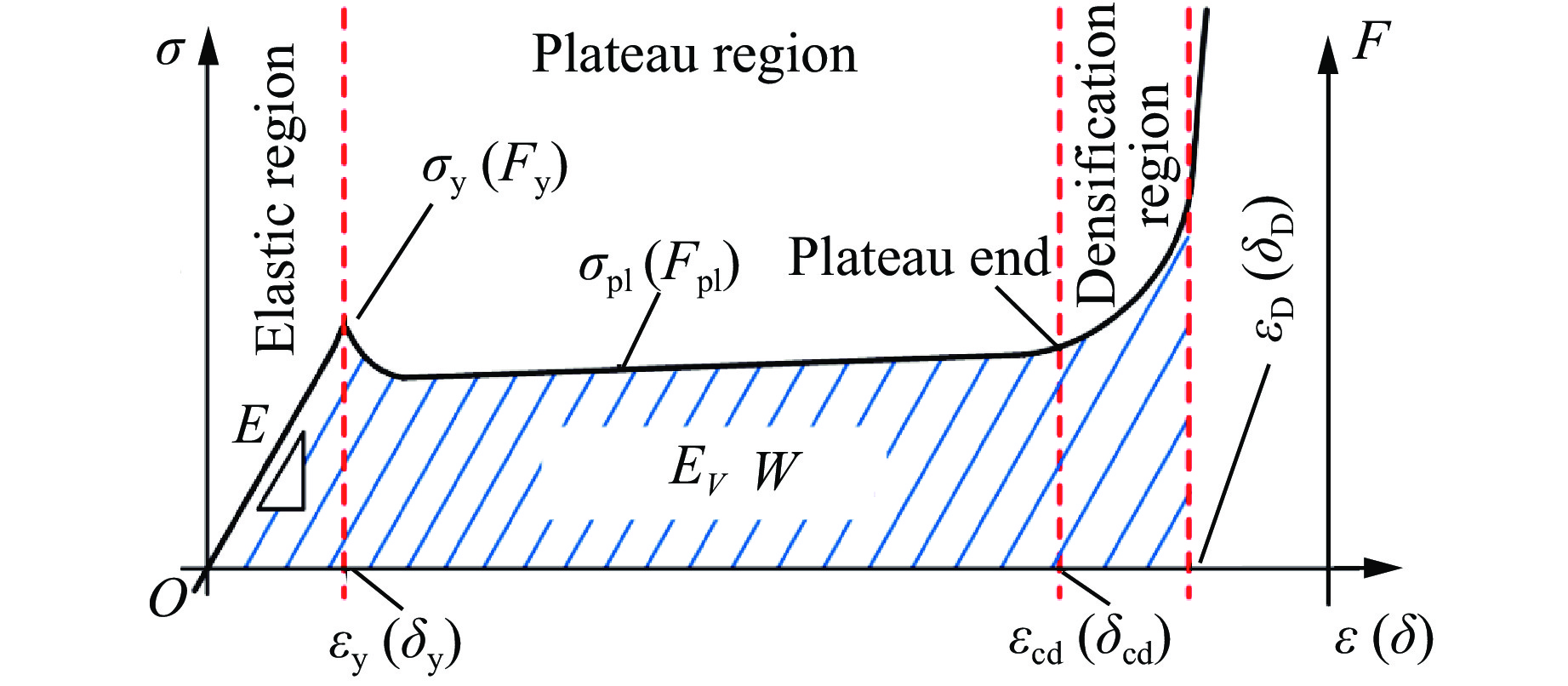
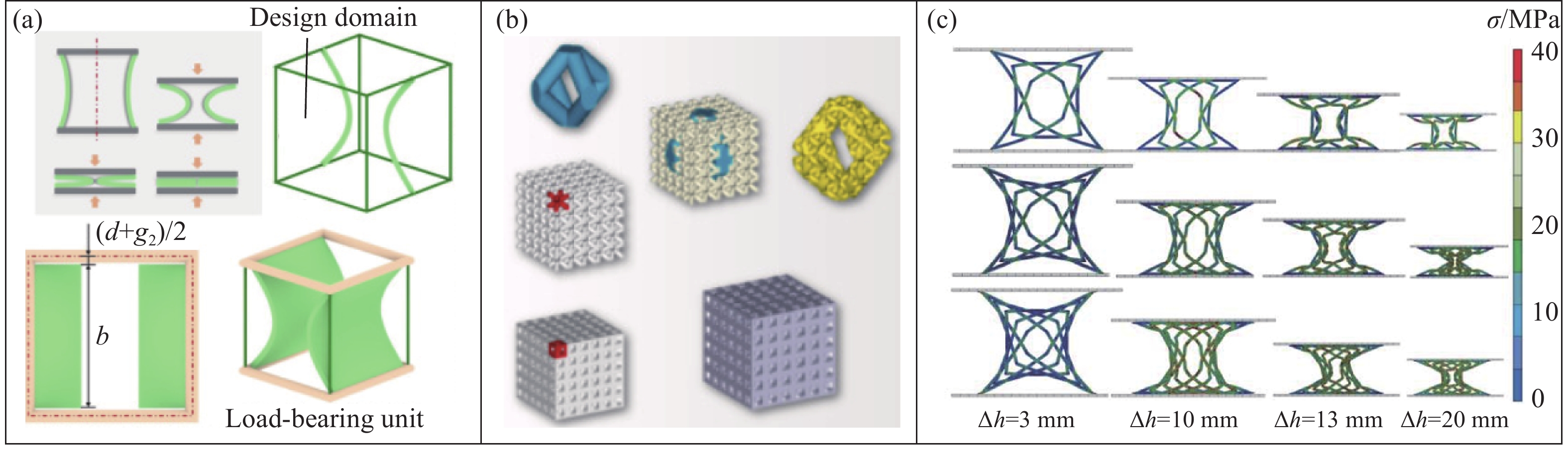
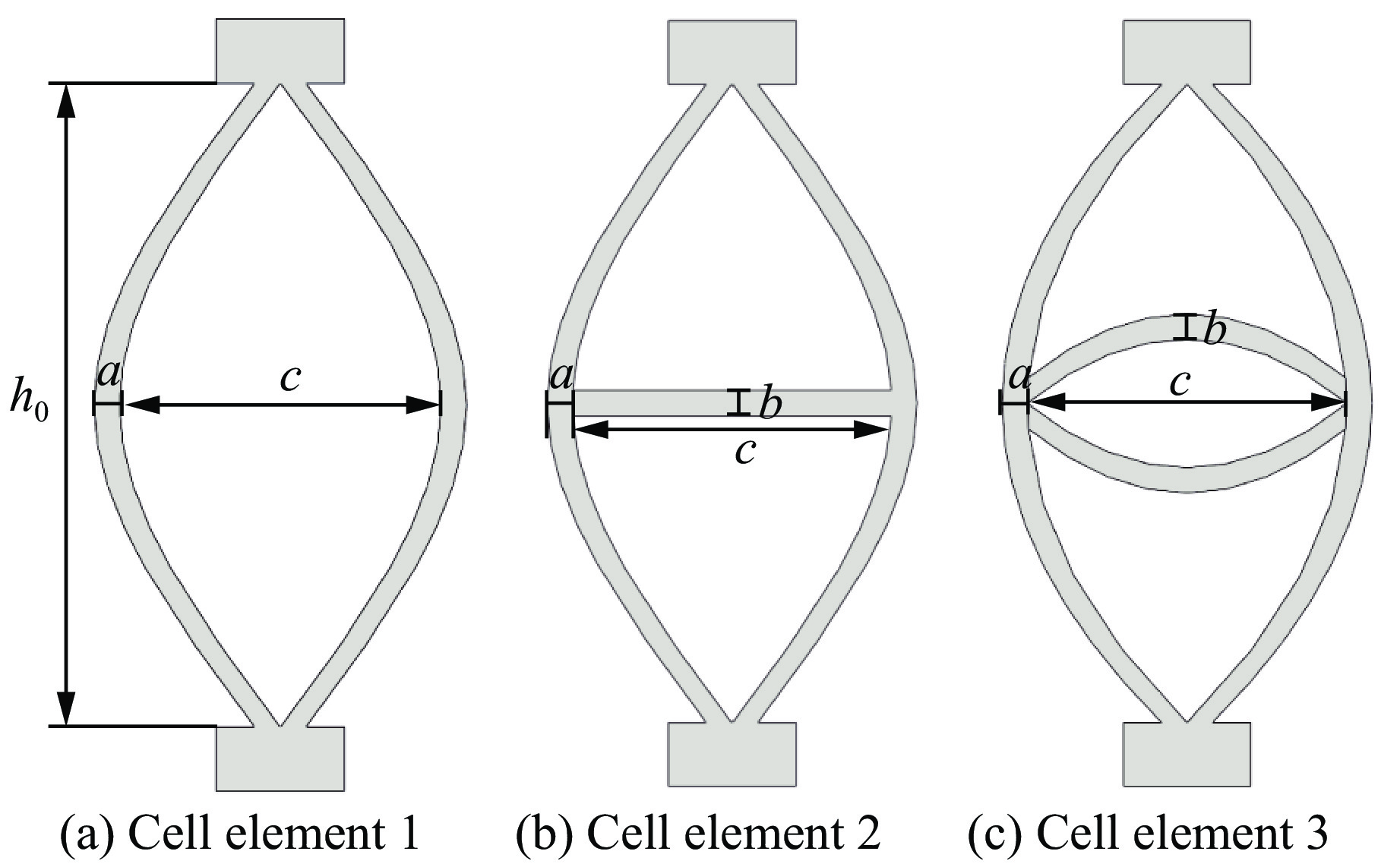
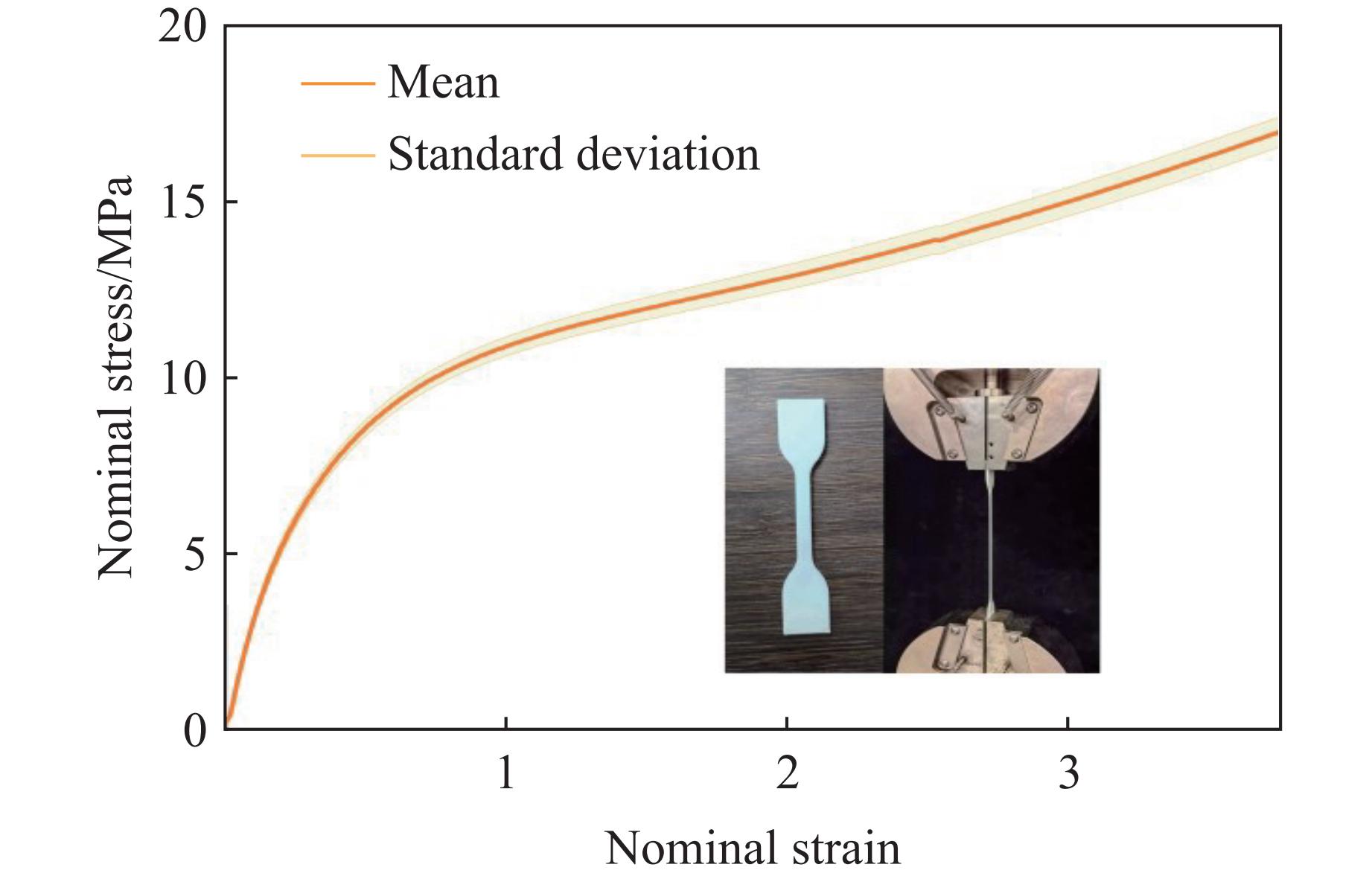
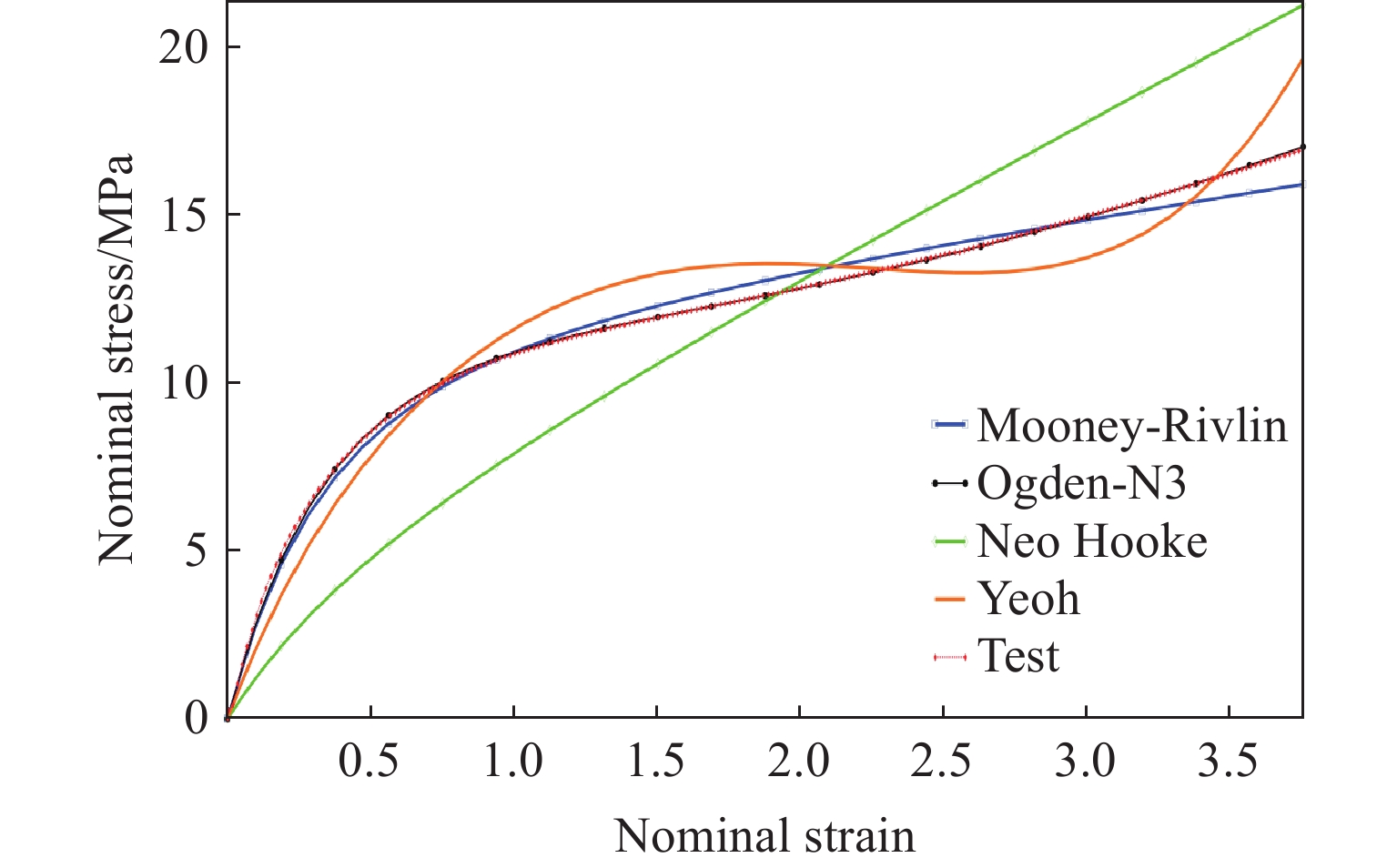
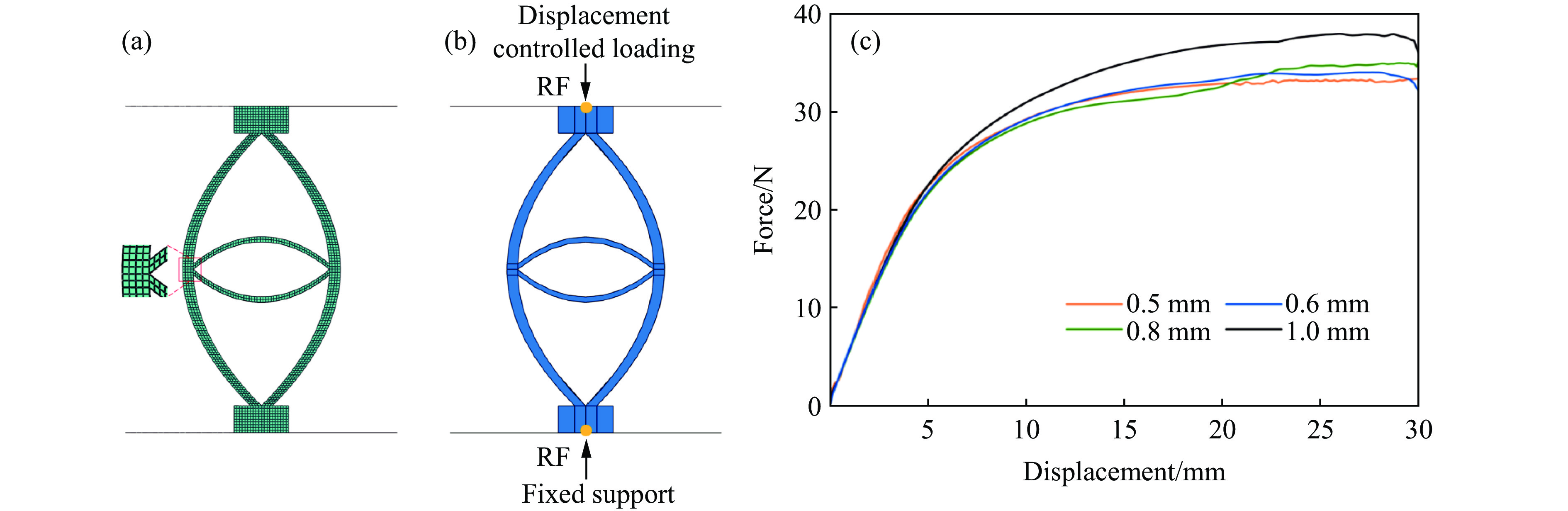
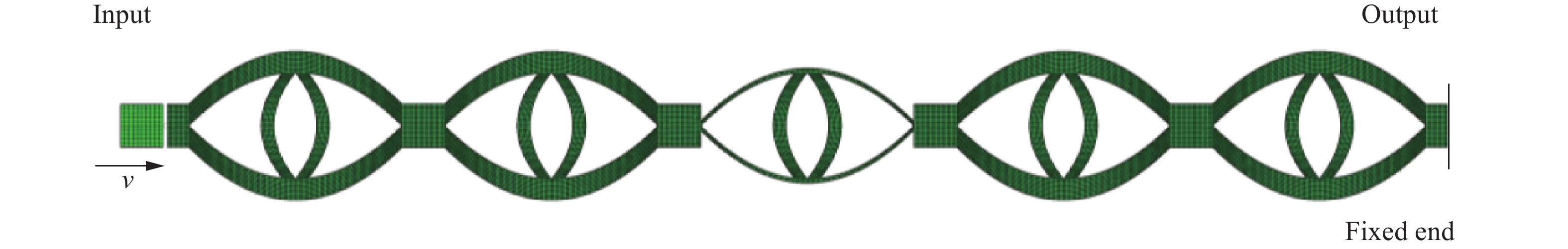
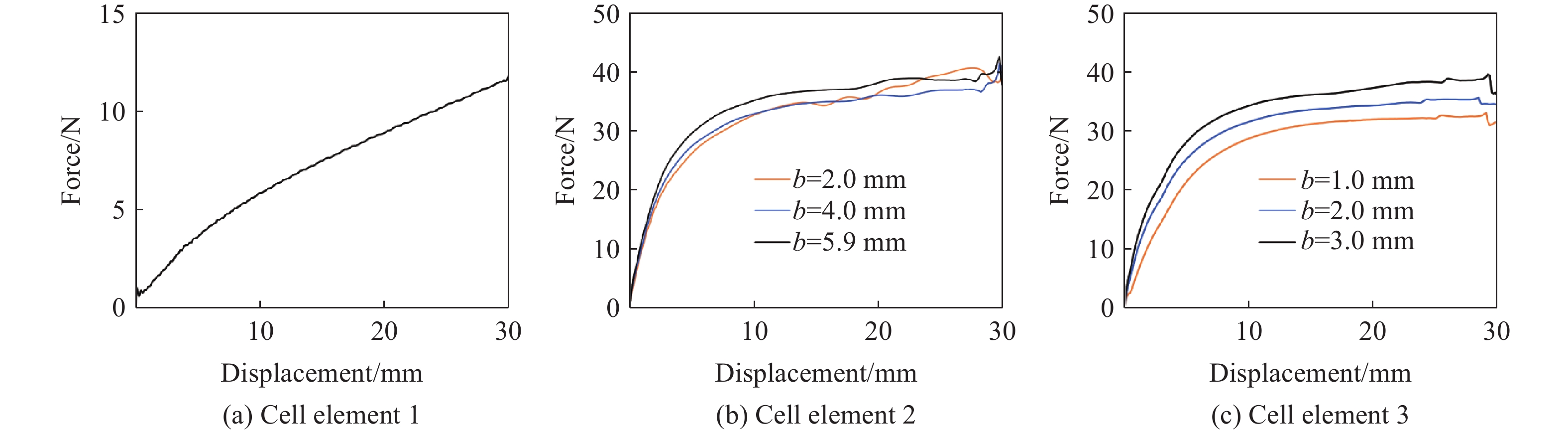
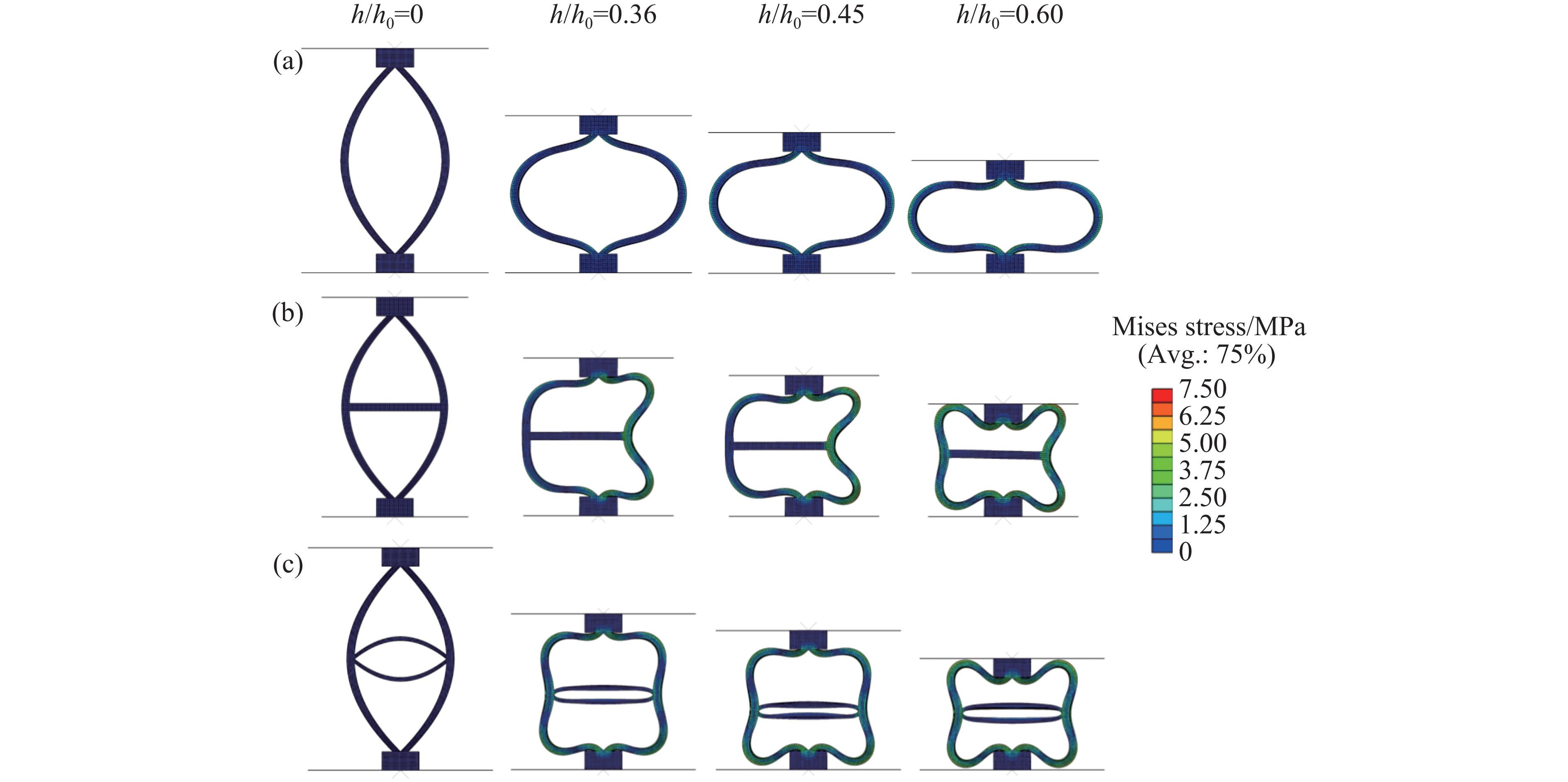
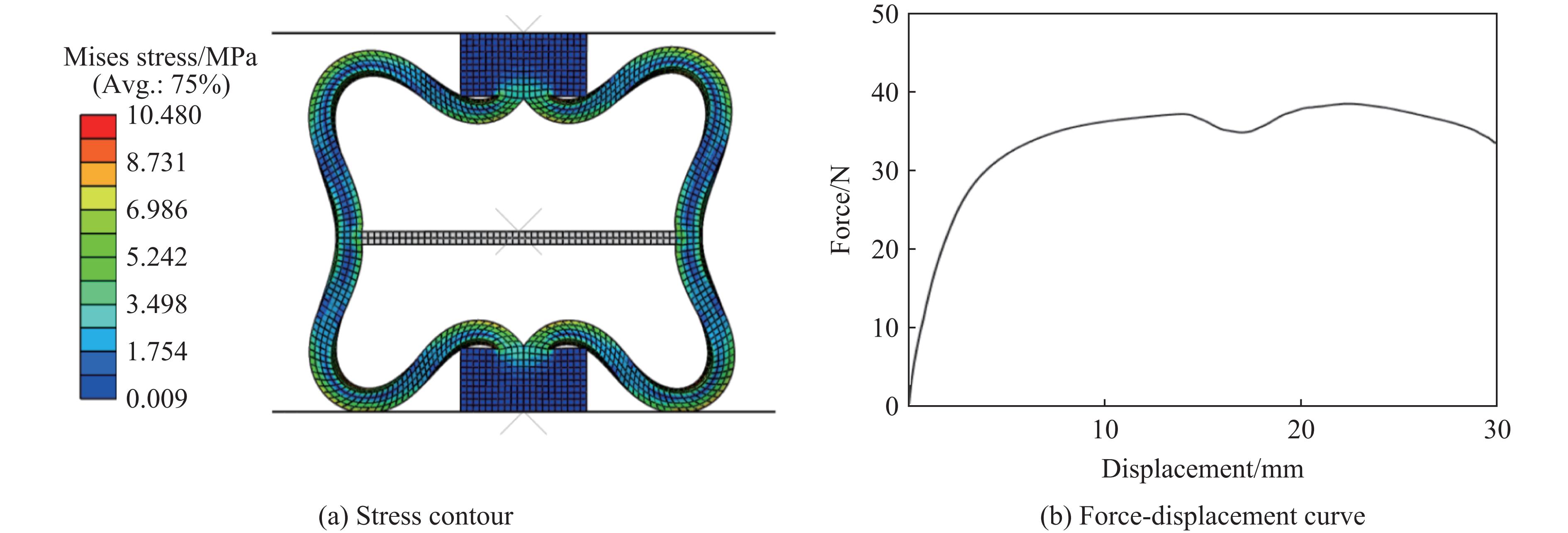
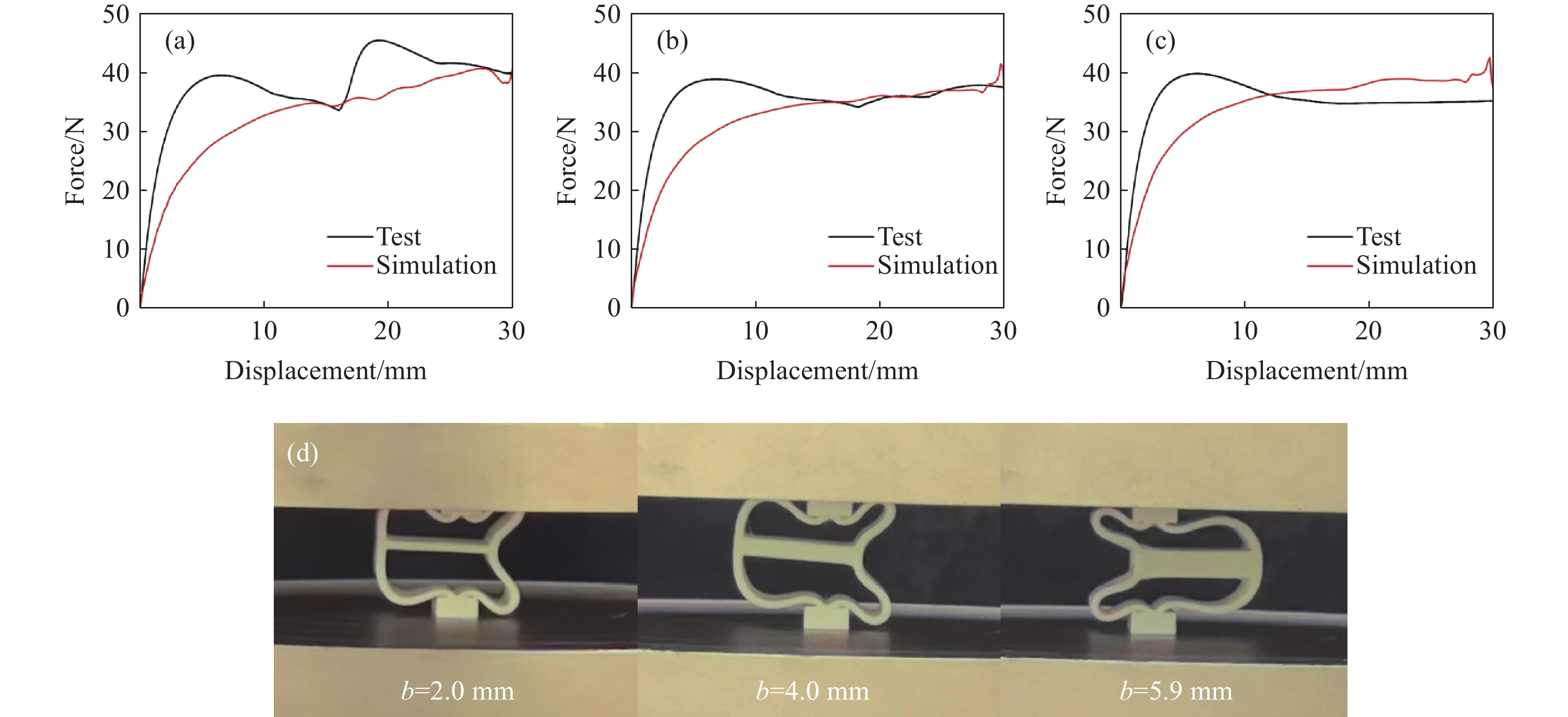
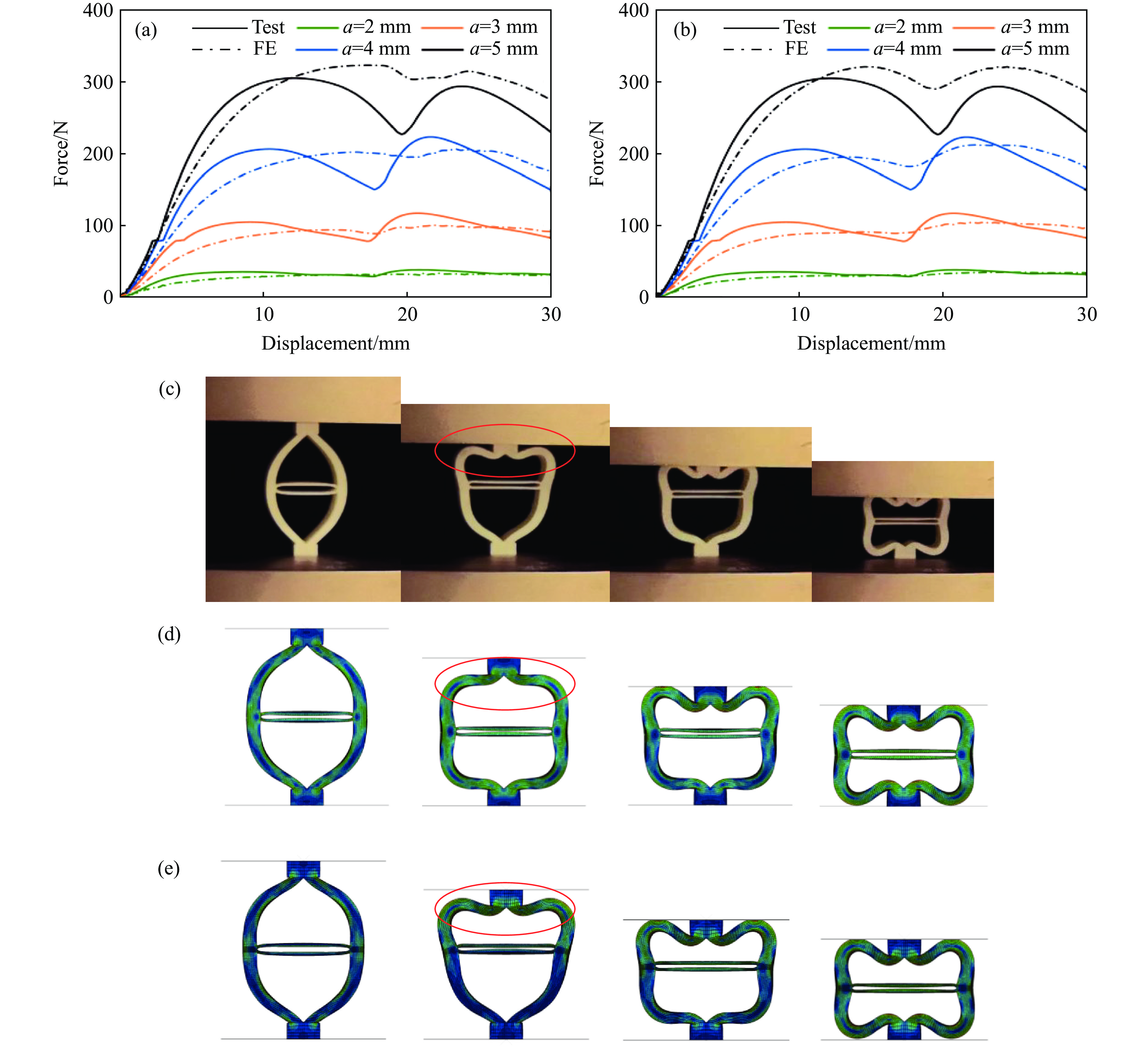
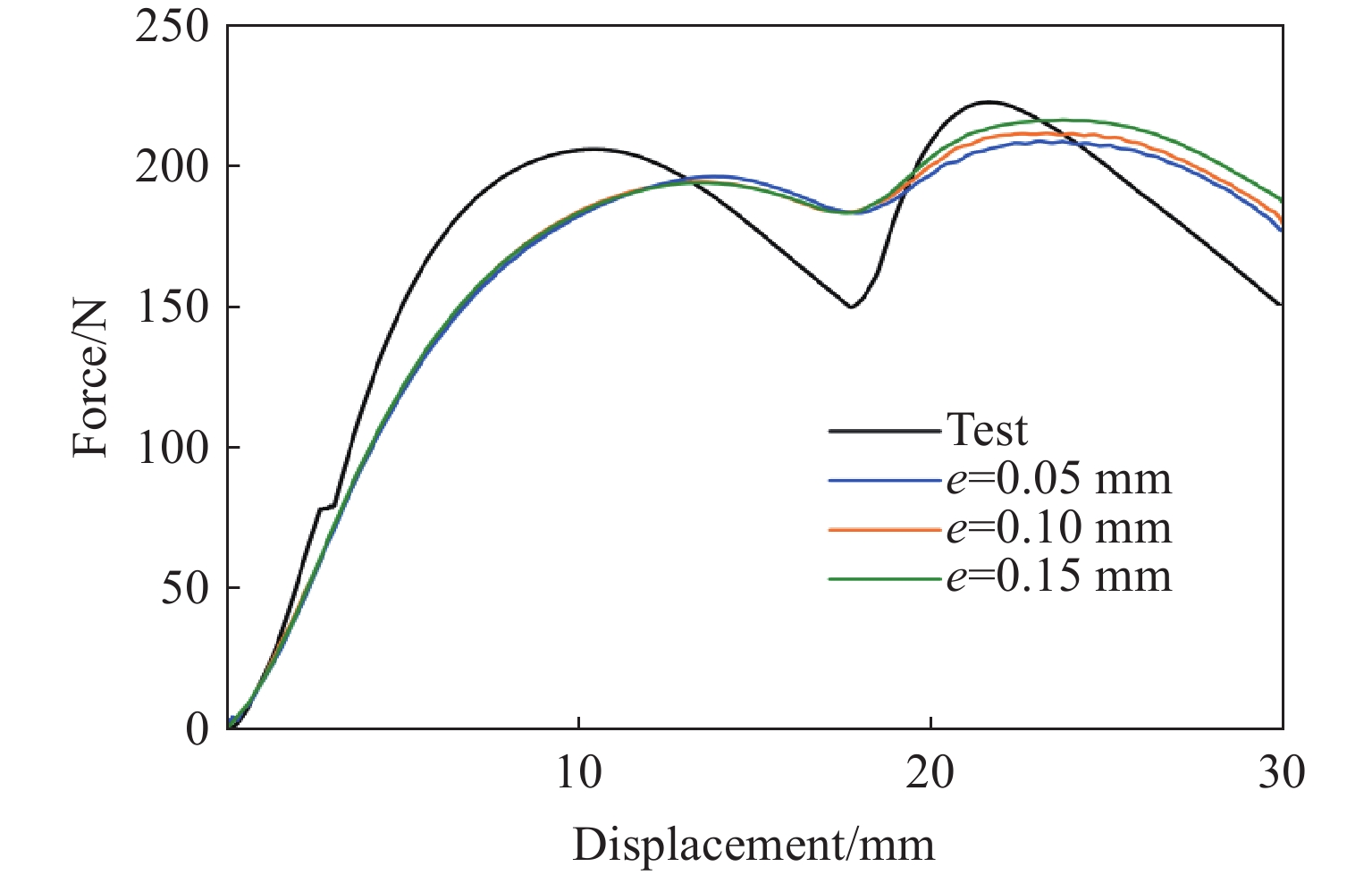
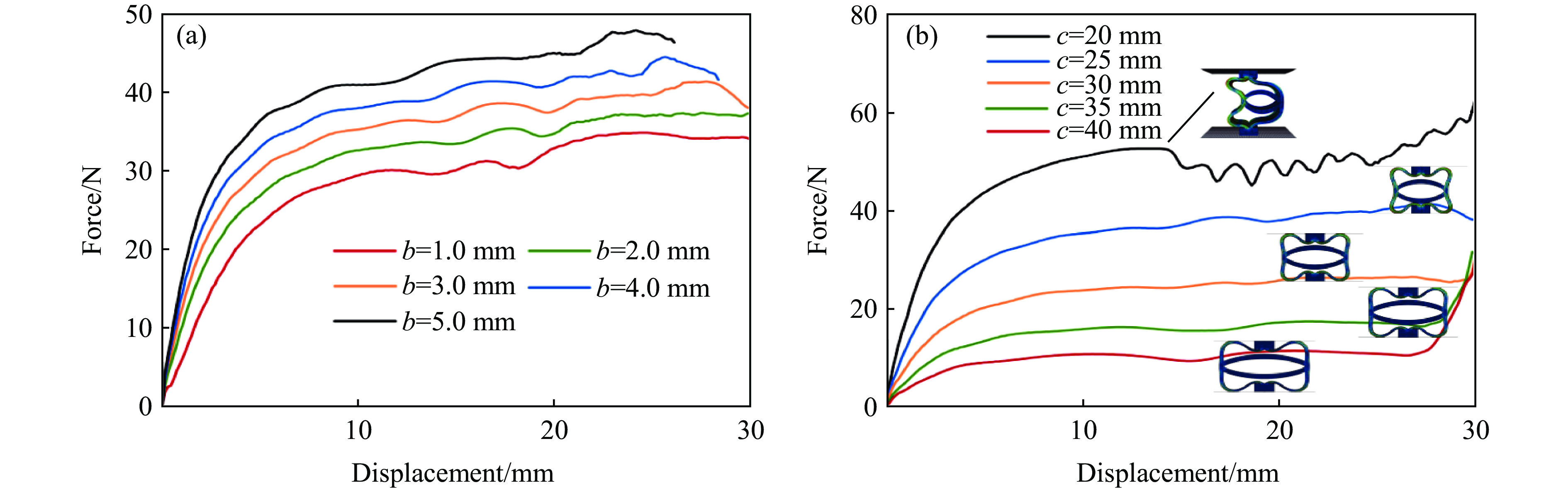
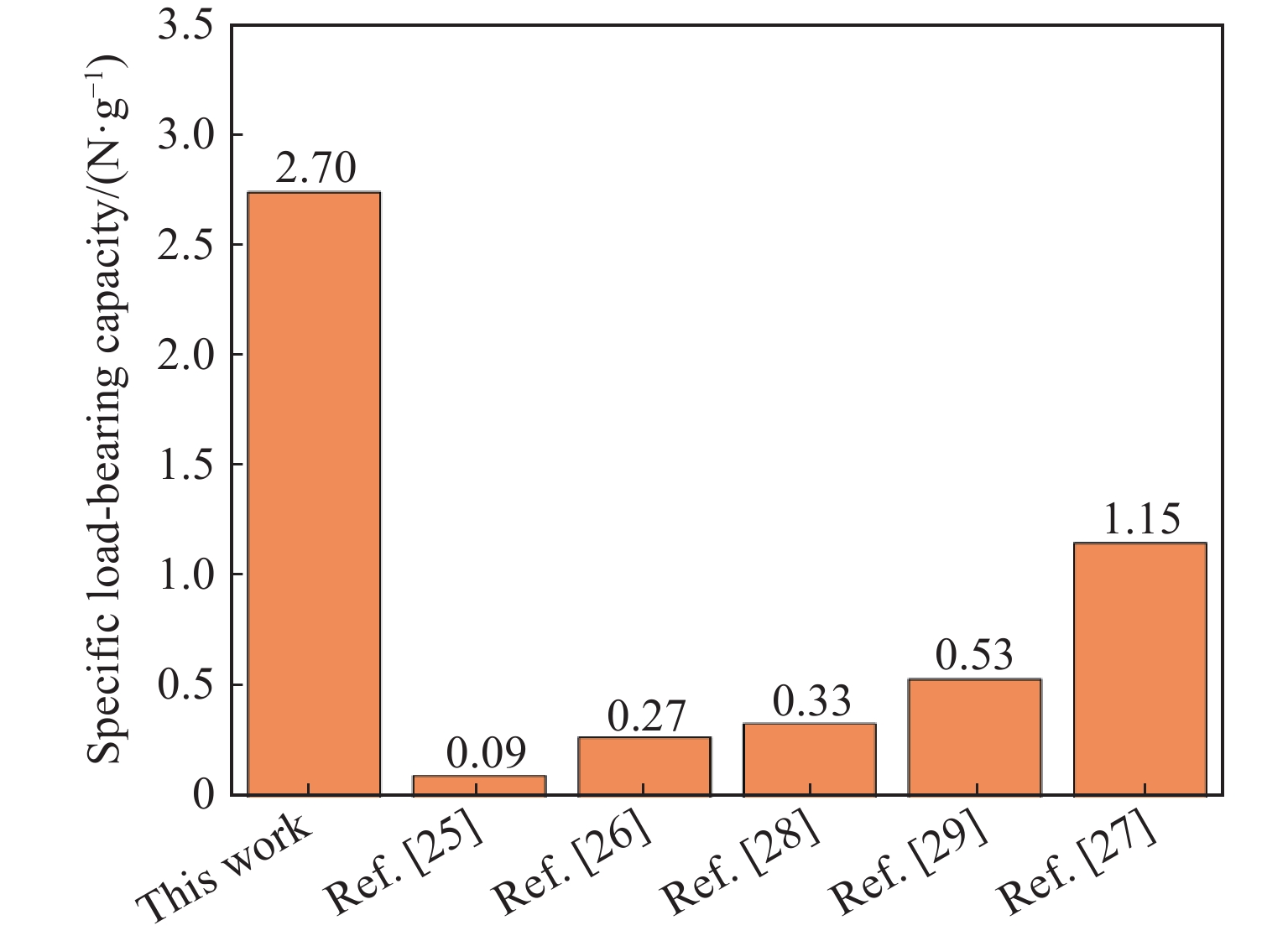
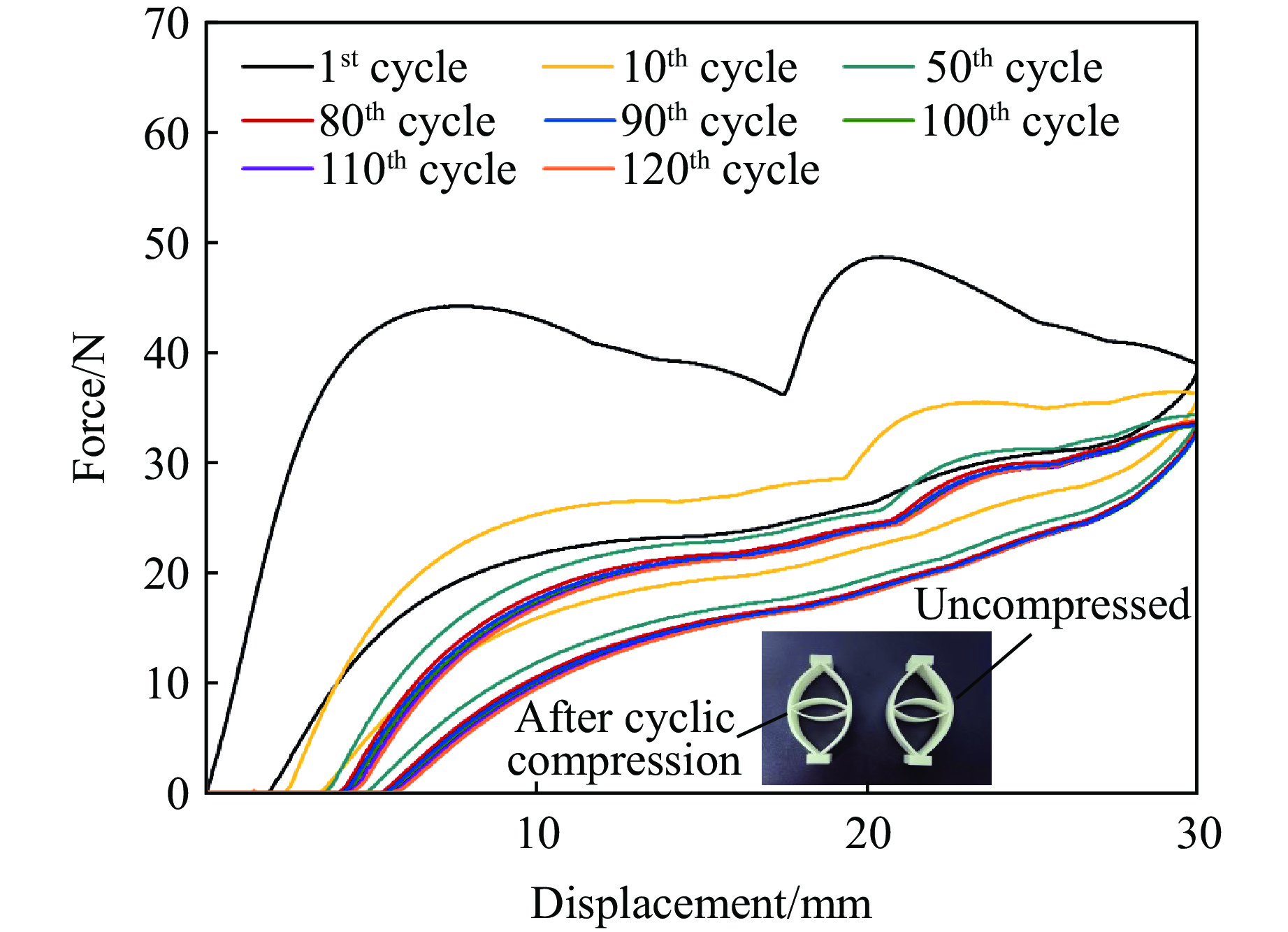
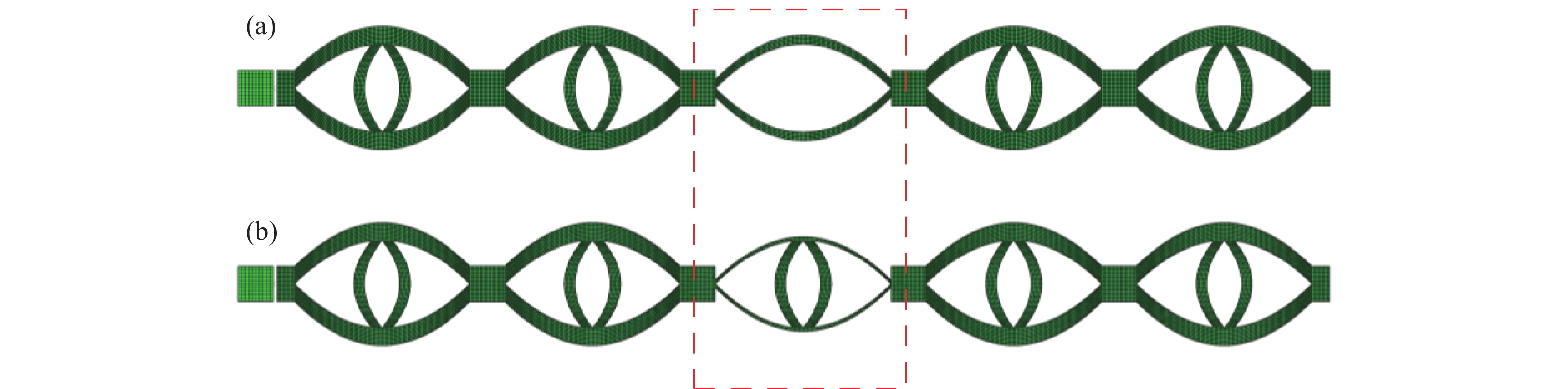
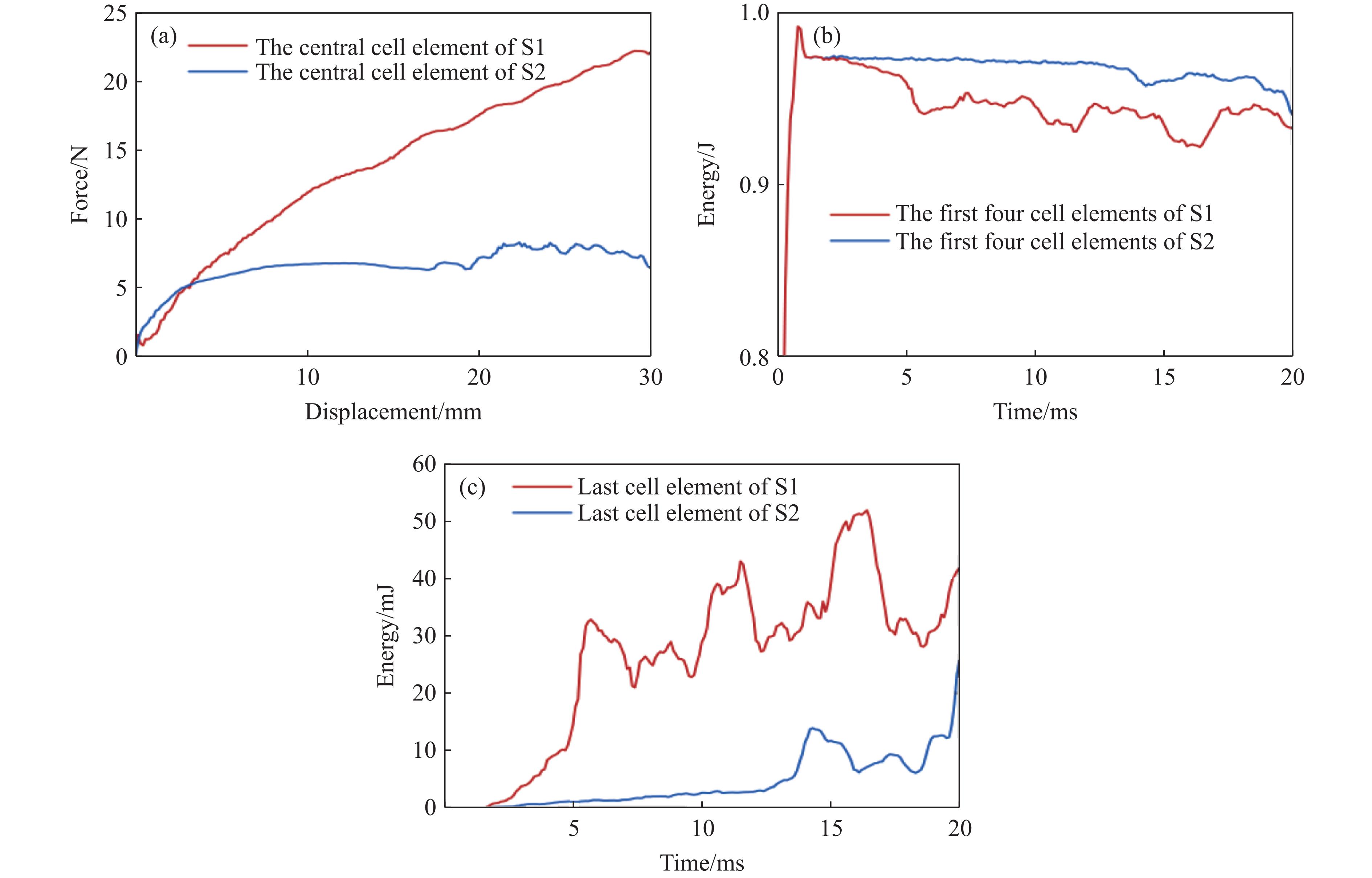
Metamaterials with both reversible deformation and a long load plateau meet the demand for cyclic buffering, offering great application prospects in protective engineering. However, current metamaterials generally suffer from low material utilization, which limits their load-bearing and energy absorption performance. To address these limitations, a novel buffering metamaterial with a long load plateau is proposed in this work. Composed of bilaterally symmetric double-arc structures and vertically symmetric curved plates, the metamaterial is capable of recoverable large deformation and overall cooperative load-bearing deformation, thereby improving material utilization and optimizing structural load capacity and energy absorption performance. Experimental tests and numerical simulations were conducted to validate the long load plateau and recoverable large deformation characteristics of the metamaterial. The influences of structural geometric parameters on its mechanical behavior were also systematically analyzed. The results indicate that the long load plateau can be effectively tuned by adjusting the thickness of lateral double arcs, the thickness of intermediate curved plates, and the central transverse span. Once the intermediate curved plates are removed, the long load plateau feature disappears, and the force-displacement curve presents an approximately linear variation. Finite element simulations at equal mass confirm that the proposed metamaterial possesses better buffering performance than similar structures without a long load plateau, and the underlying buffering mechanism is clarified. The findings provide a novel design strategy for improving the performance of metamaterials with a long load plateau, and facilitate their application in protective engineering.


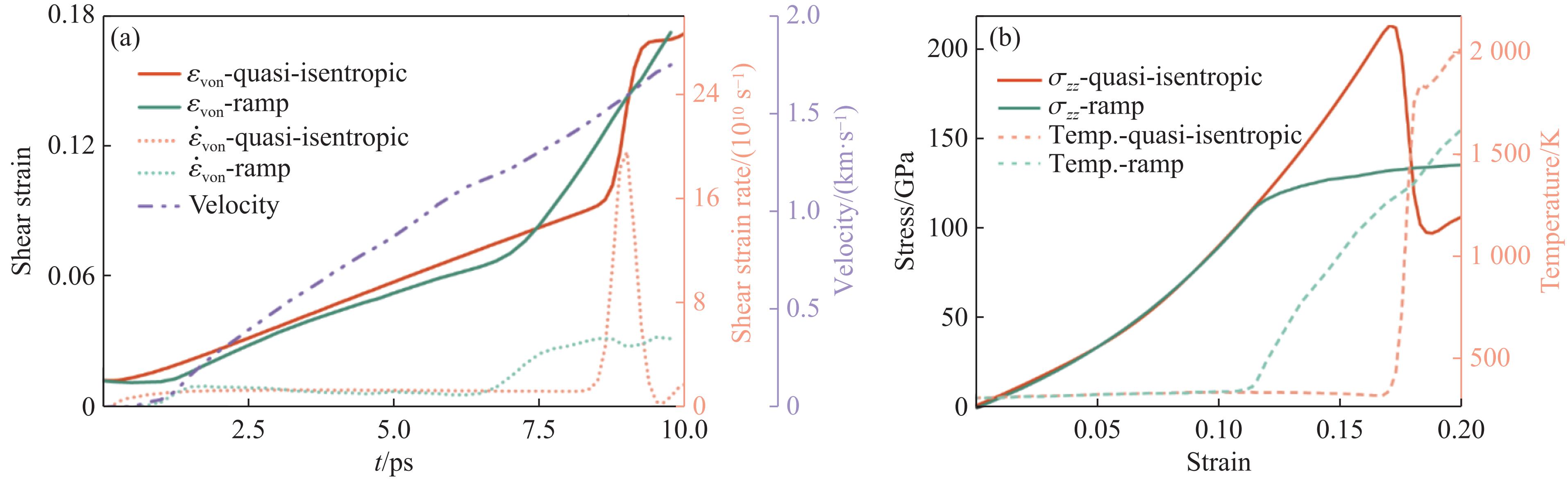
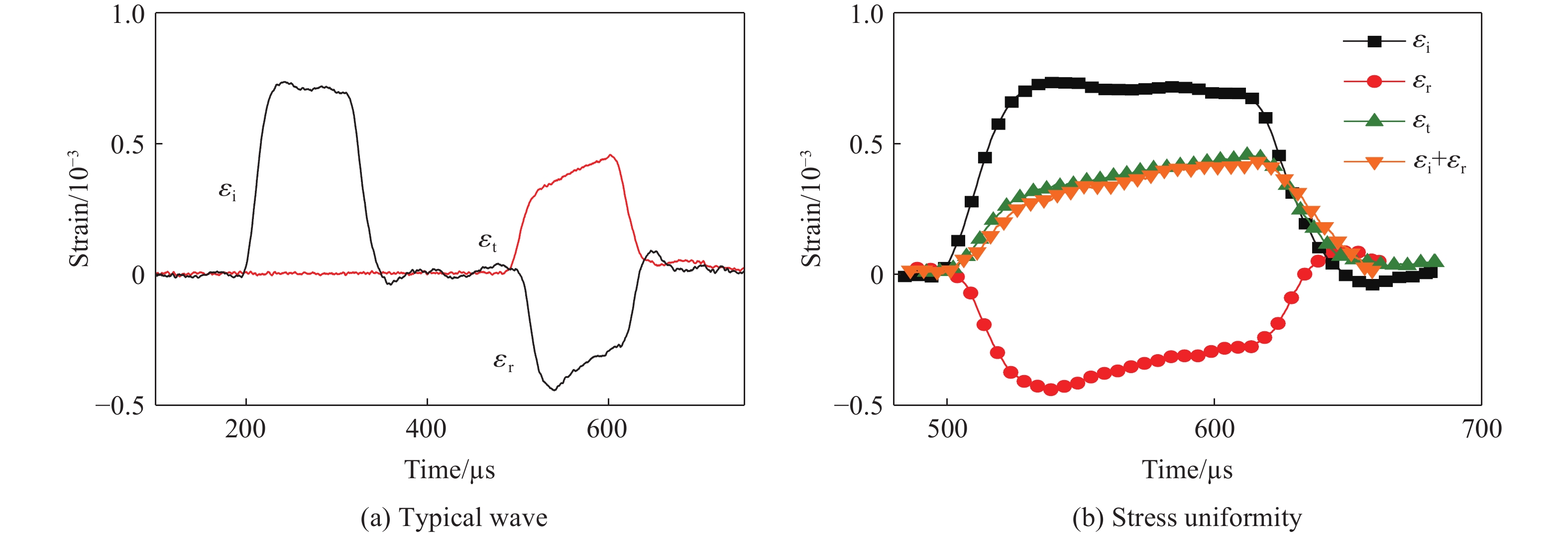
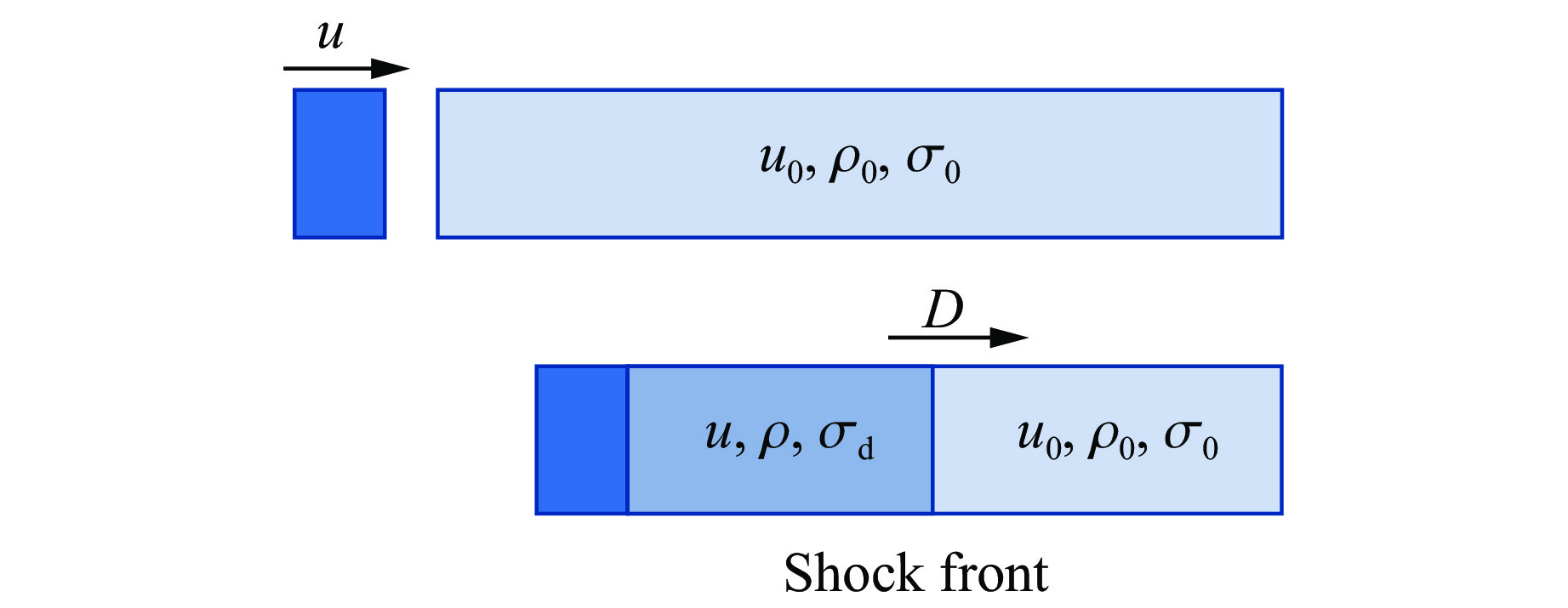
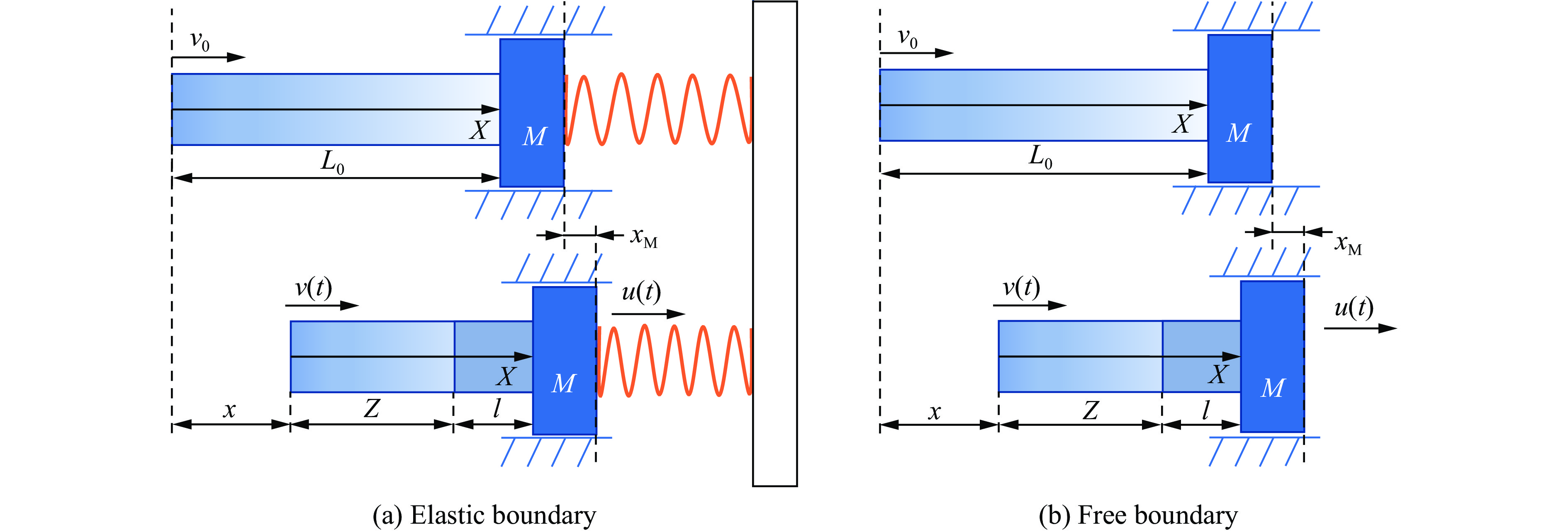
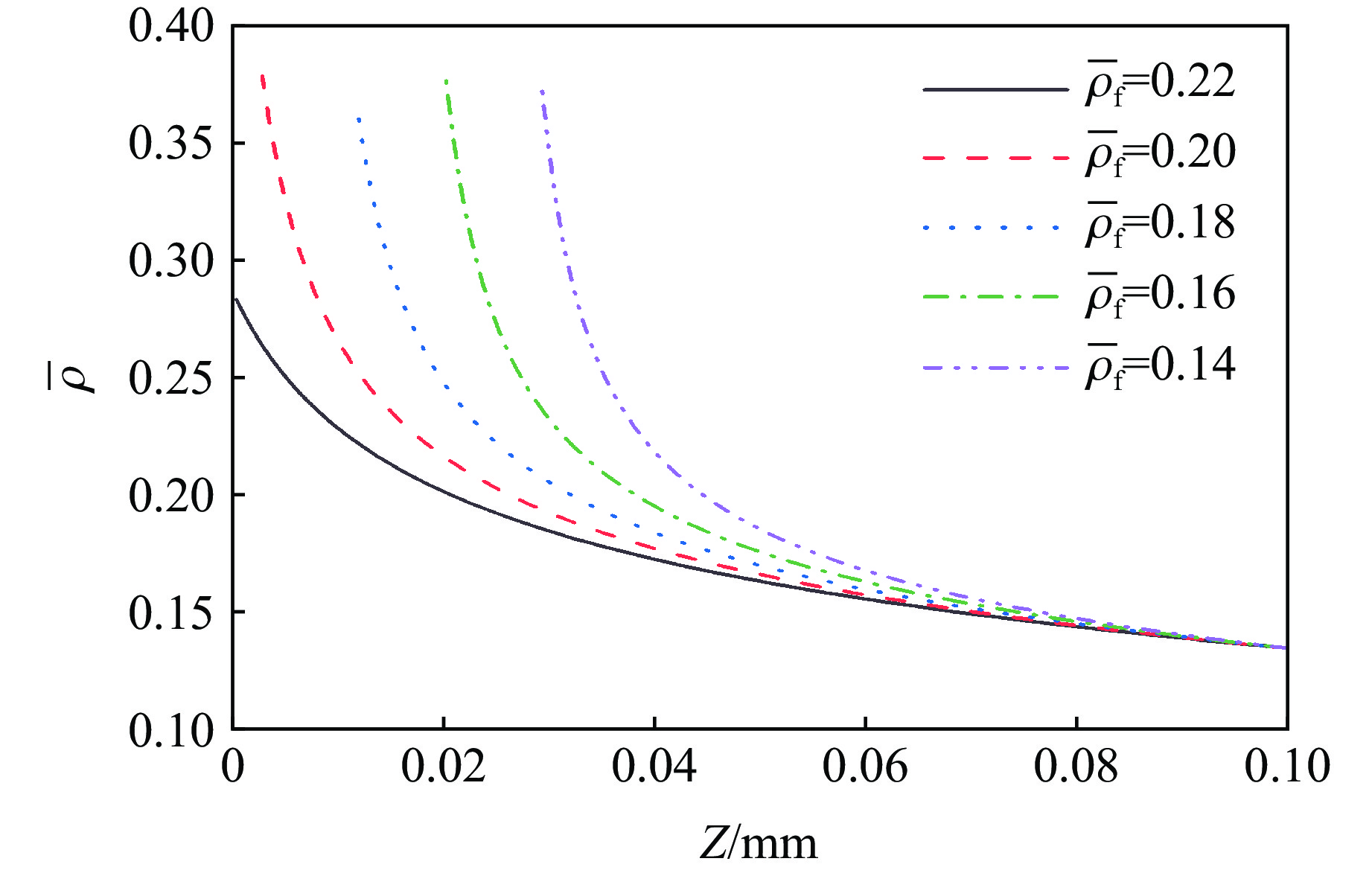
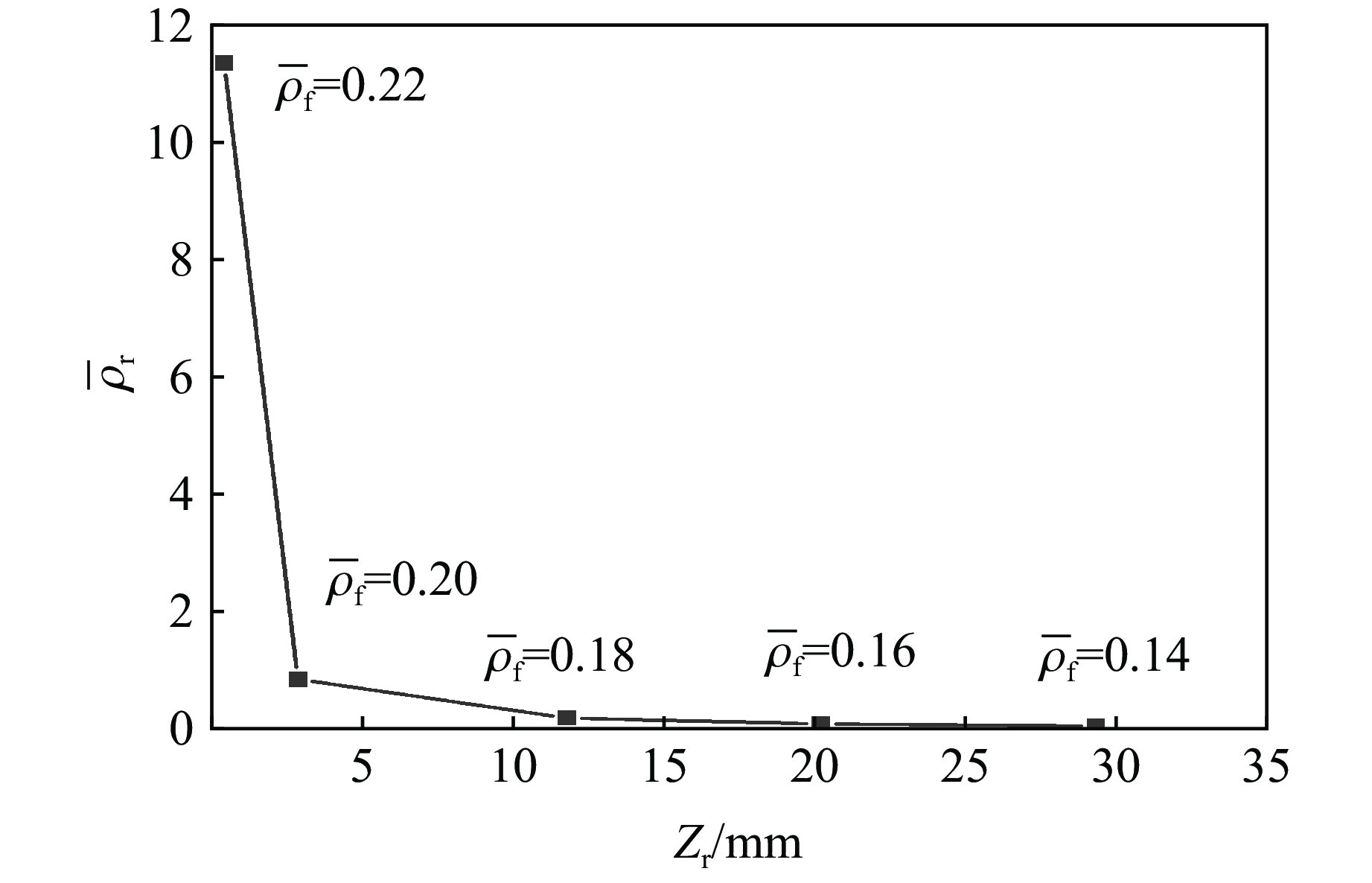
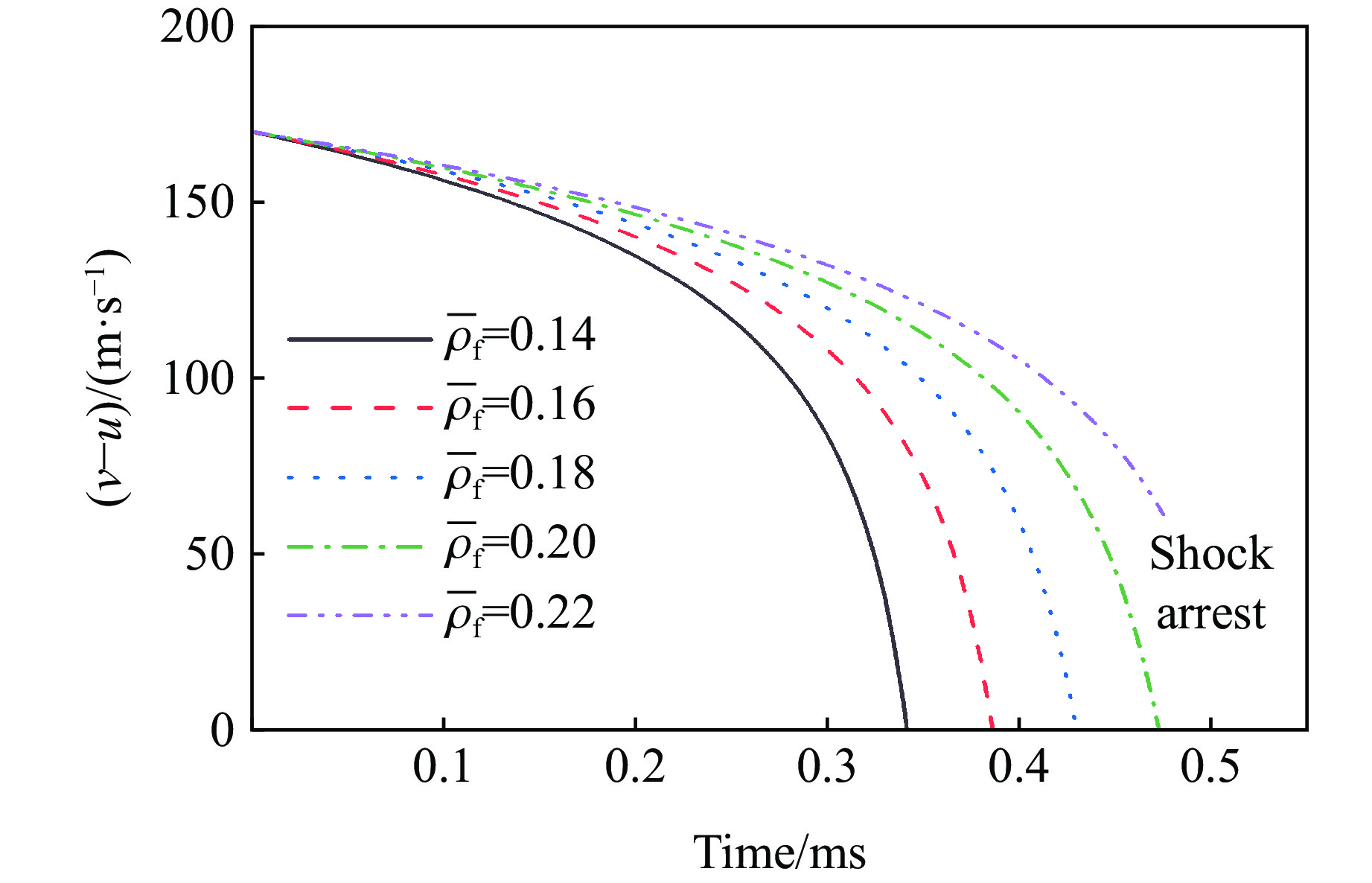
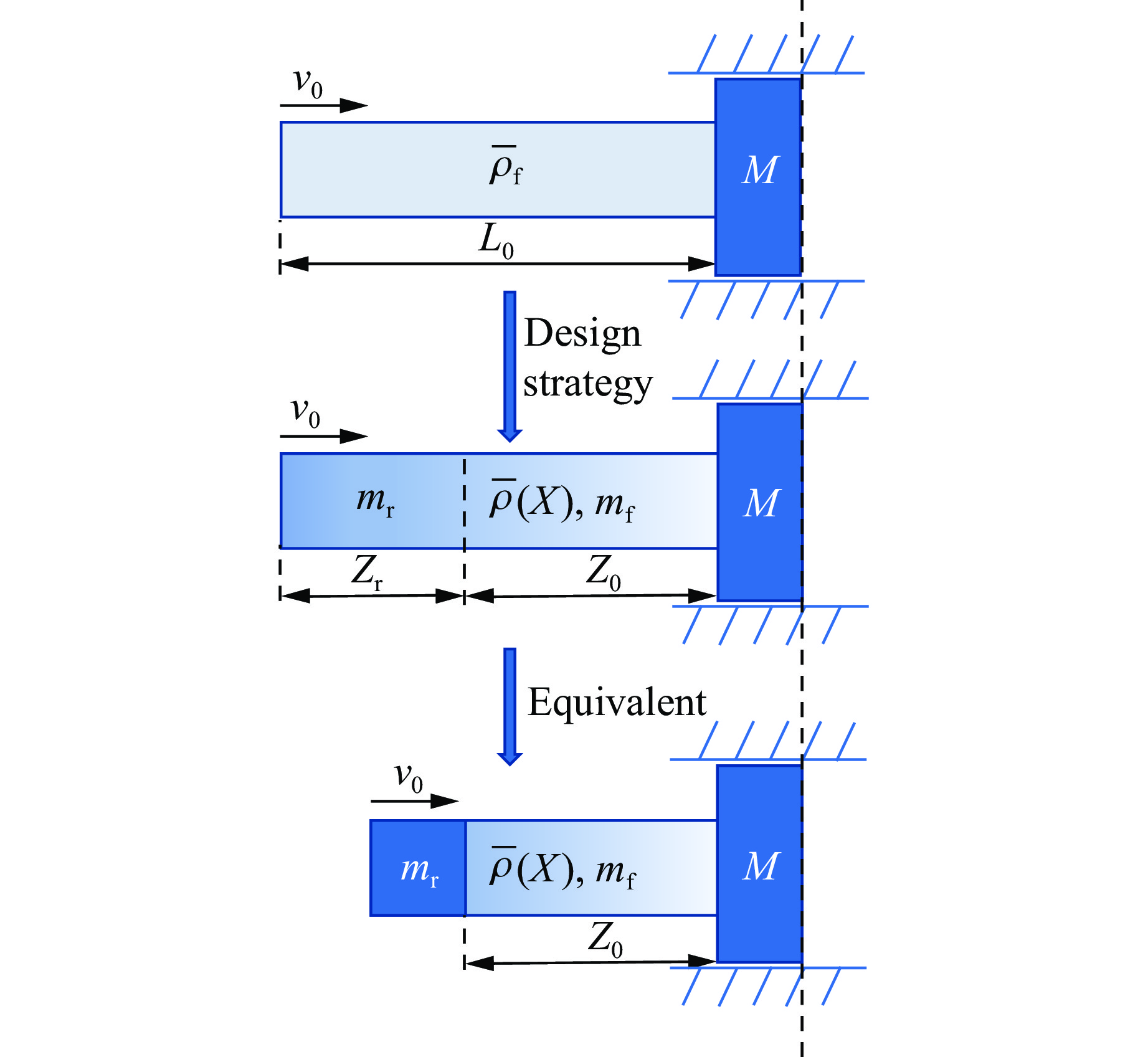
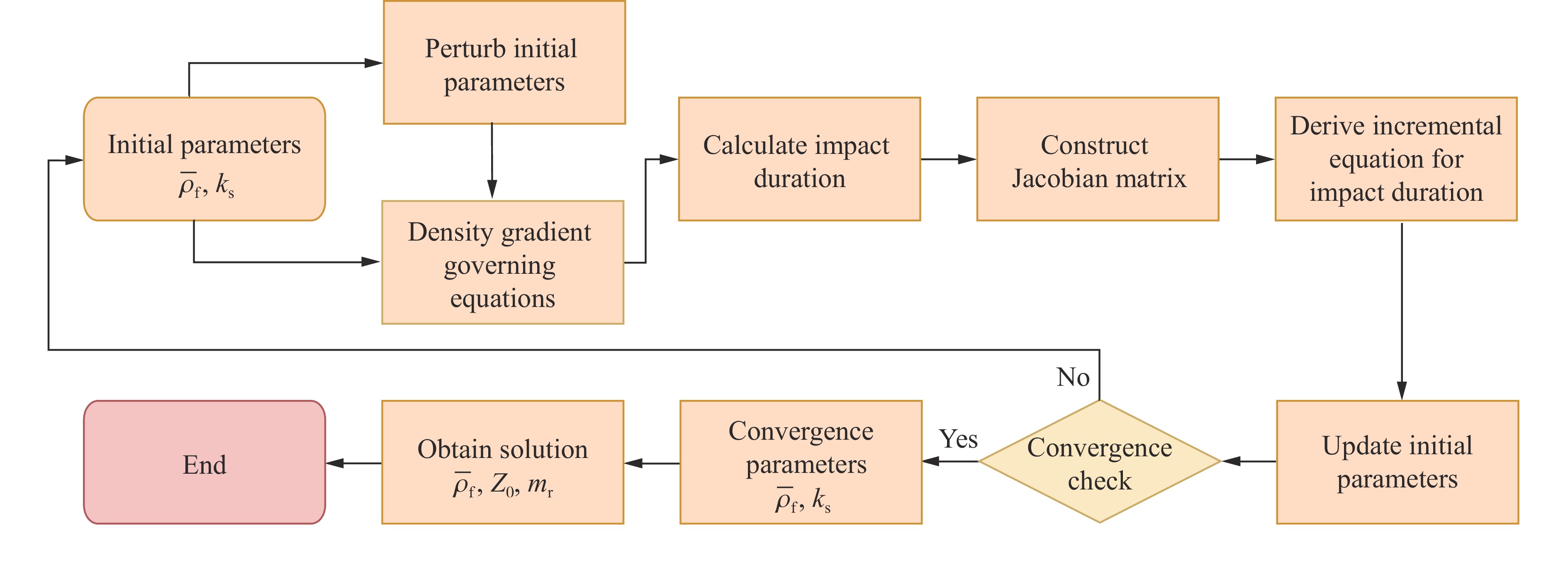
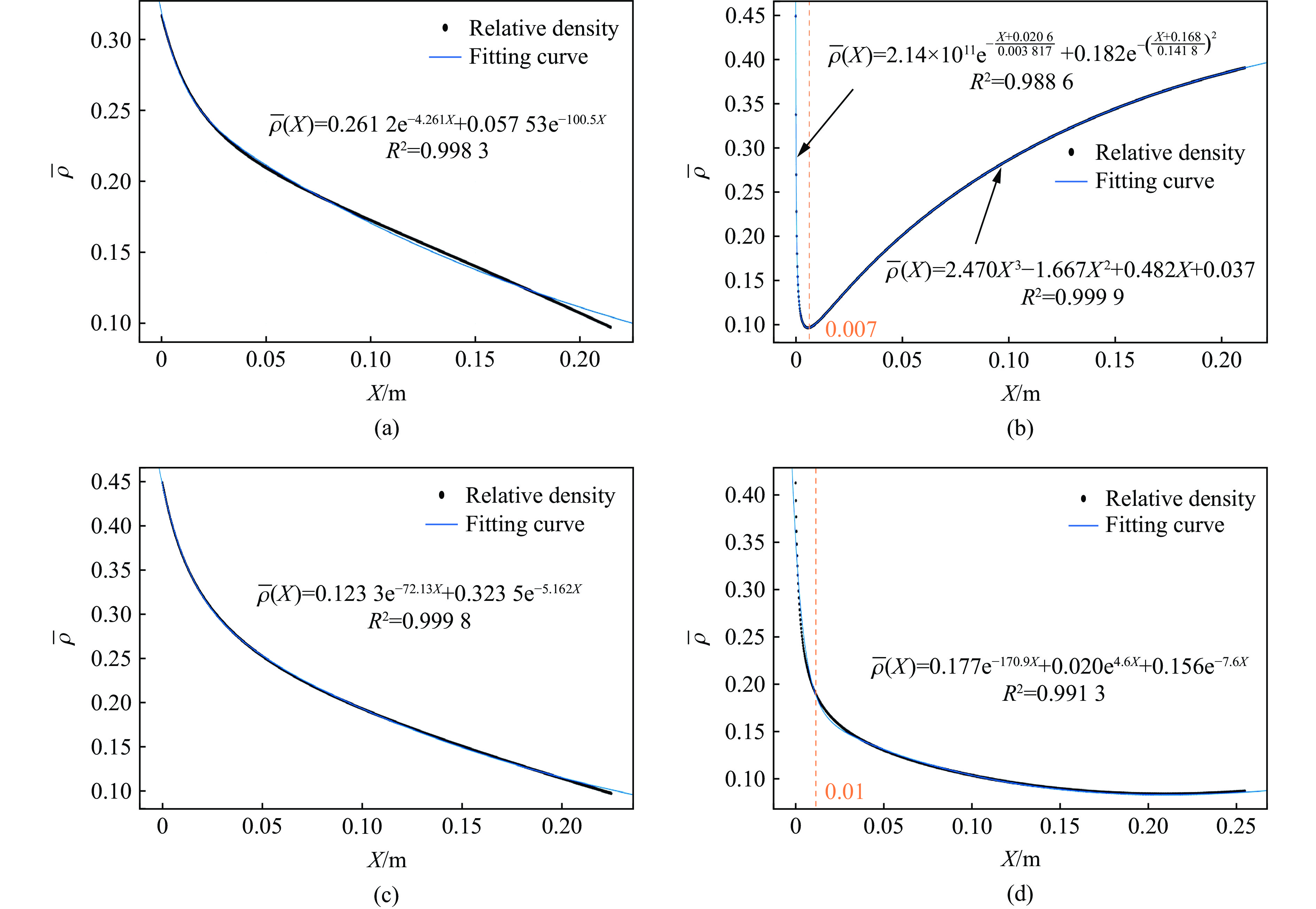
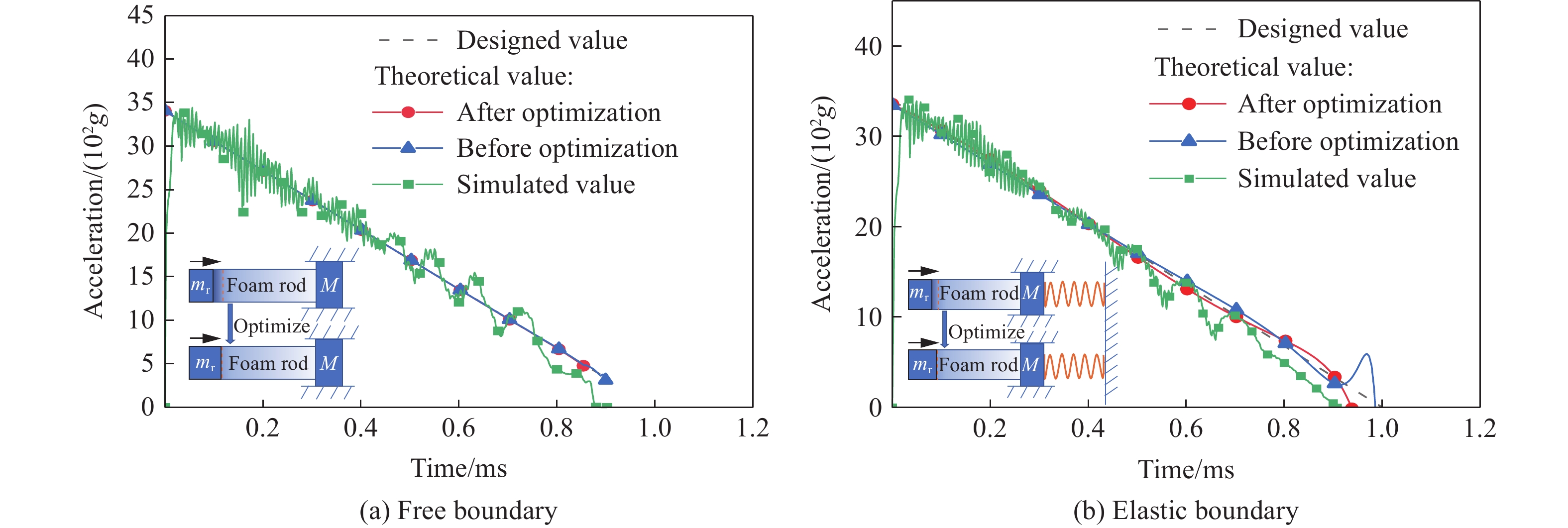
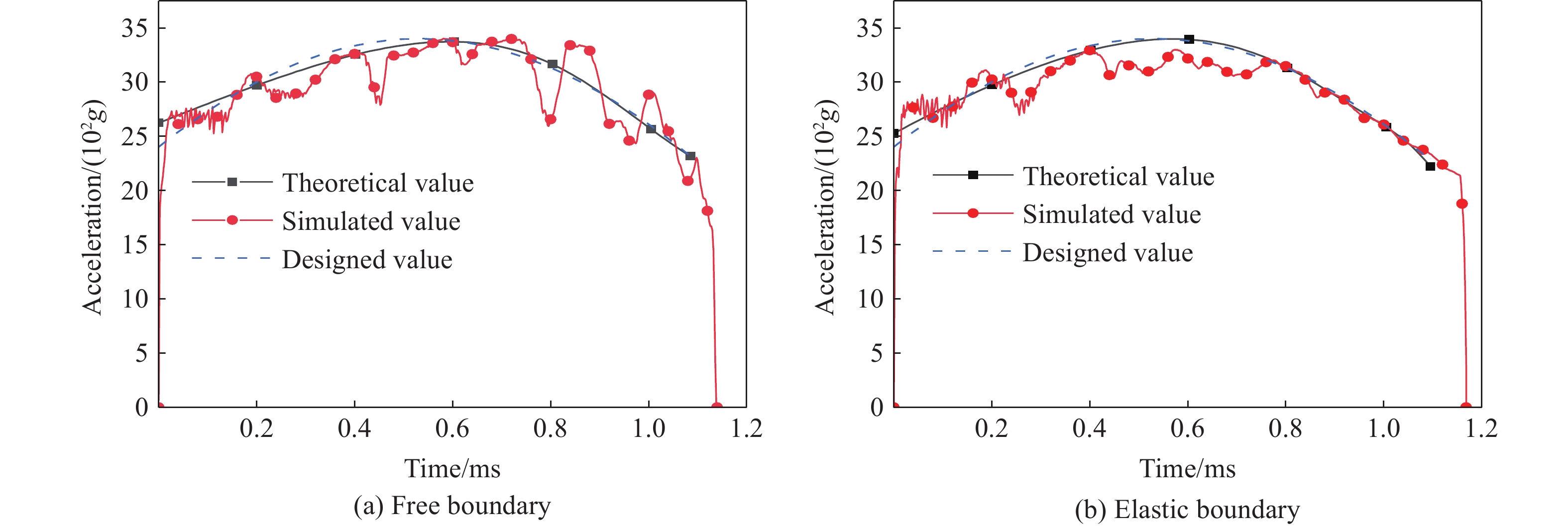
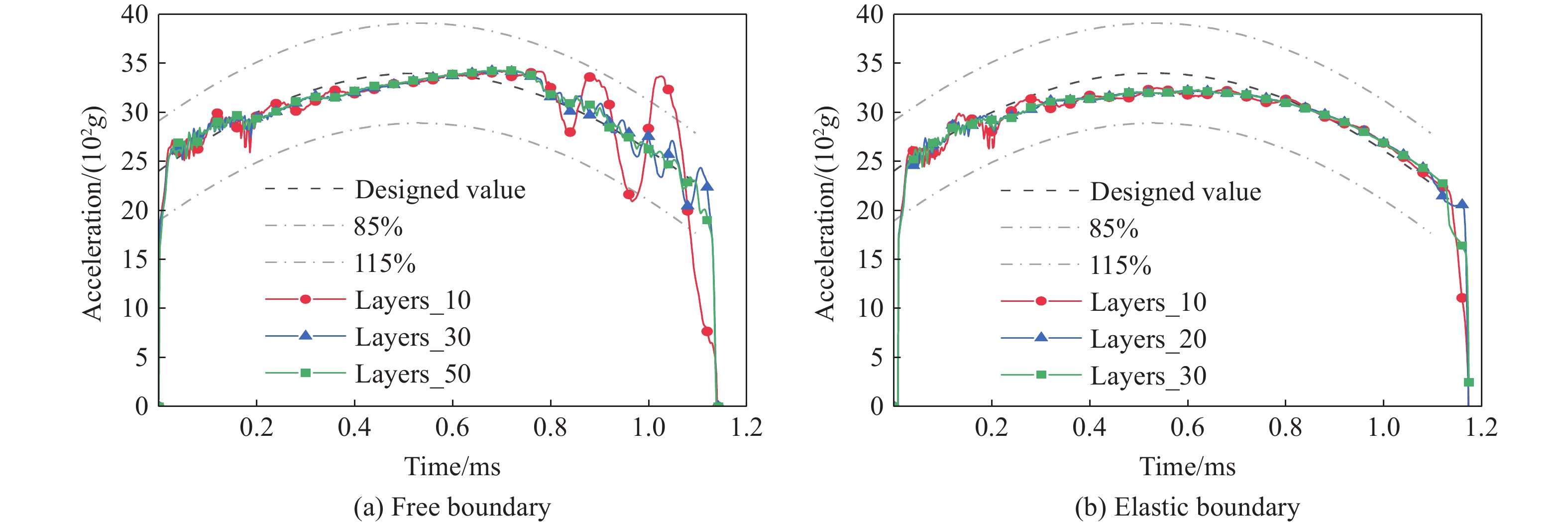
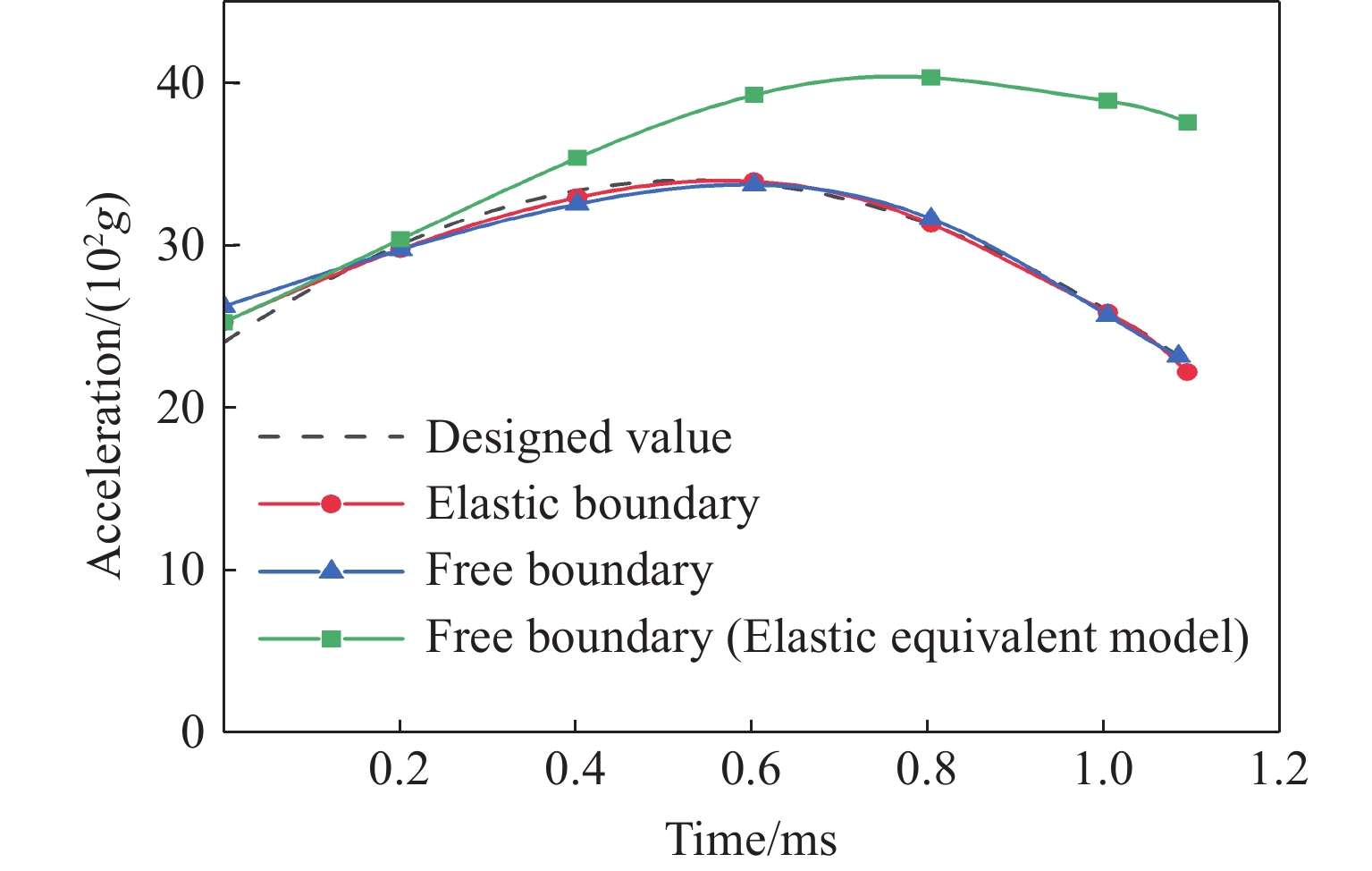
To achieve the precise generation of high-amplitude impact waveforms required for aviation safety testing and related fields, this study investigates the impact waveform regulation mechanisms of graded cellular metals under different boundary conditions. Based on the conservation laws of mass and momentum, theoretical models for impact waveform generation using graded cellular metals are established for both free and elastic boundary conditions. Furthermore, an inverse design method for density gradient is proposed, which incorporates an average relative density constraint combined with Gauss-Newton iteration, enabling the reverse solution from a prescribed acceleration waveform to the material density gradient distribution. Finite element results demonstrate that the proposed method can effectively generate required waveforms—such as triangular and half-sine waves—under both boundary conditions. The study also reveals that: free boundaries are more suitable for generating high amplitude and long duration waveforms, whereas elastic boundaries can improve the realizability of low-amplitude waveforms through stiffness regulation; boundary conditions do not alter the impact duration but exert a significant influence on waveform shape; and excessive impedance mismatch between adjacent layers will intensify waveform oscillations, thereby compromising the waveform generation accuracy. The proposed inverse design strategy for density gradients exhibits favorable versatility and provides both theoretical support and a practical design tool for the development of high-amplitude impact testing technologies.
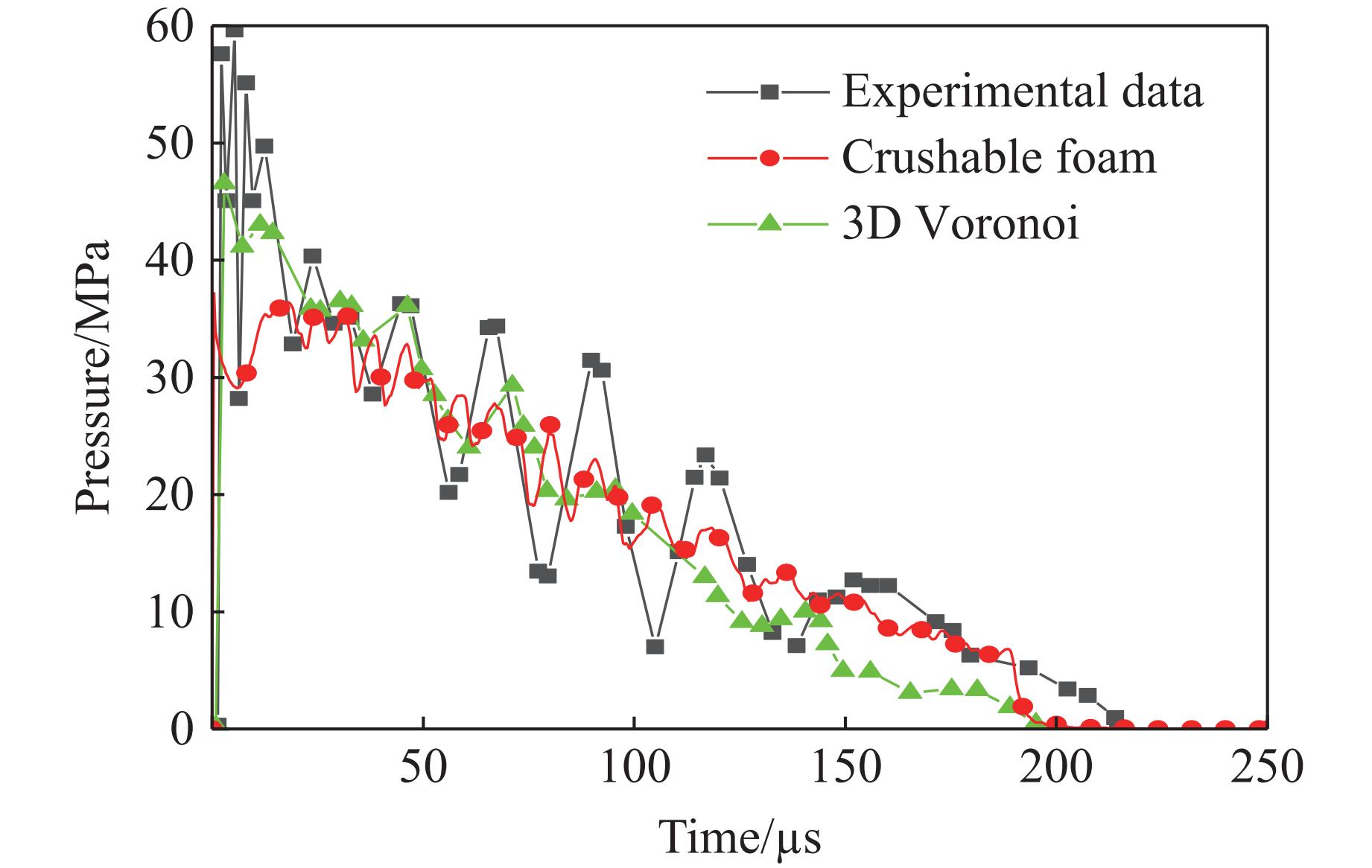
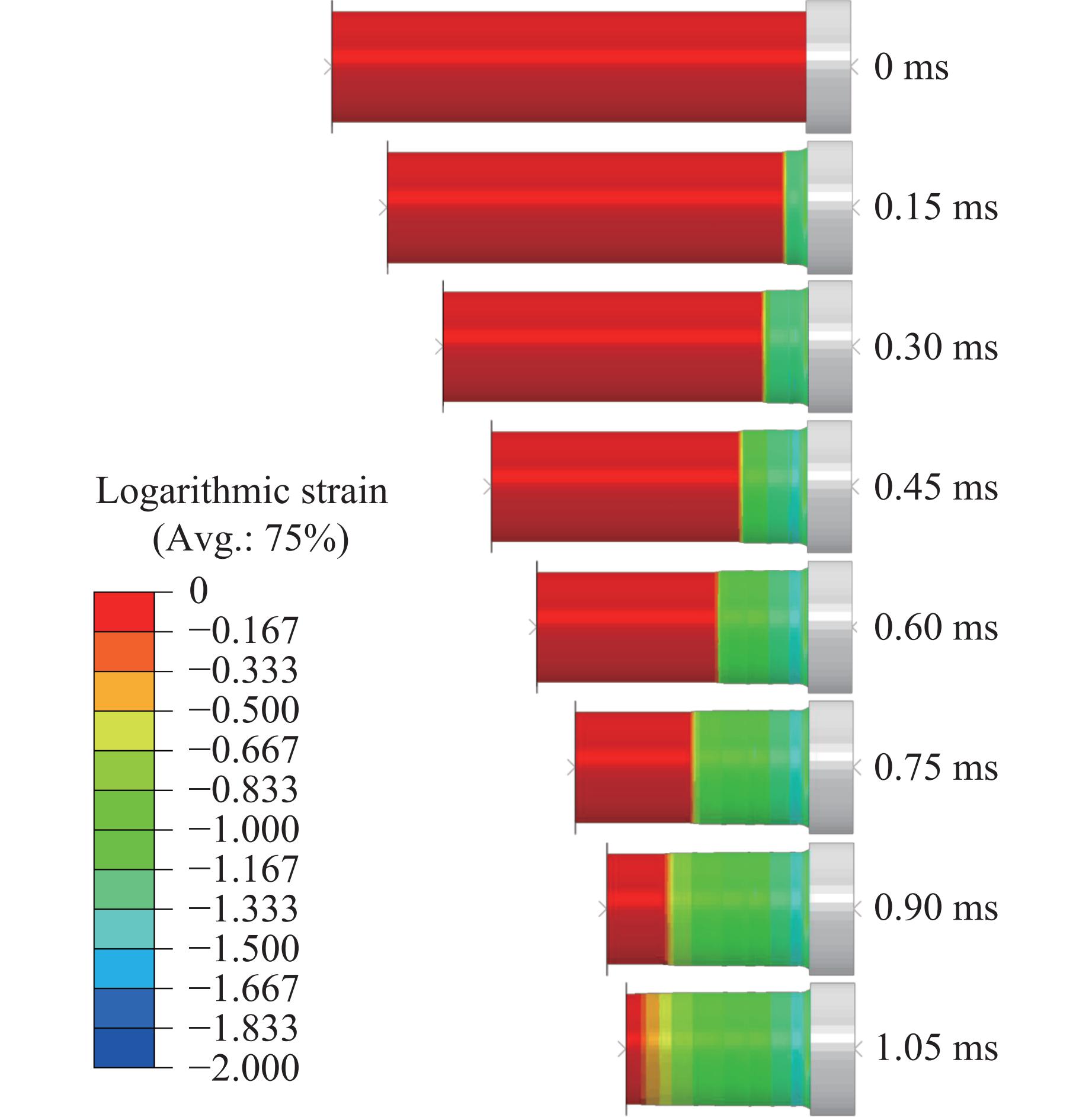
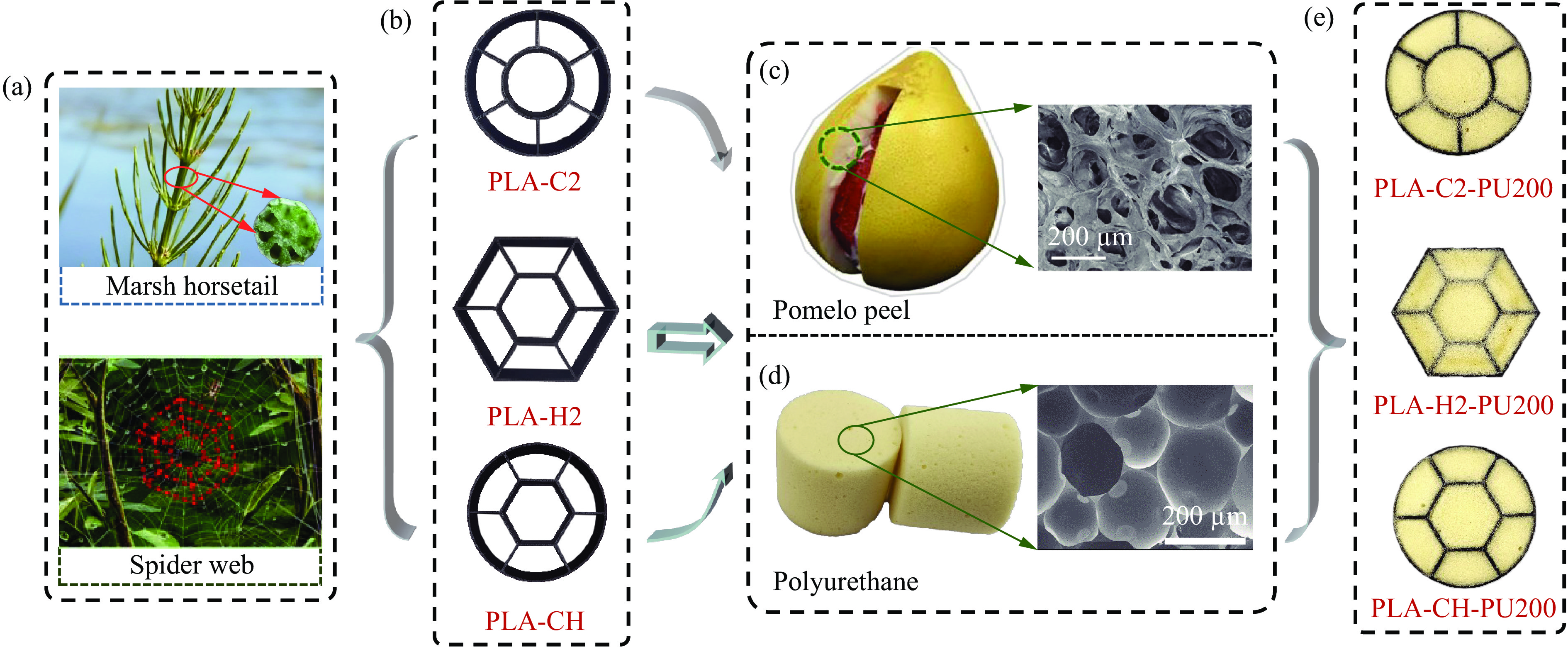
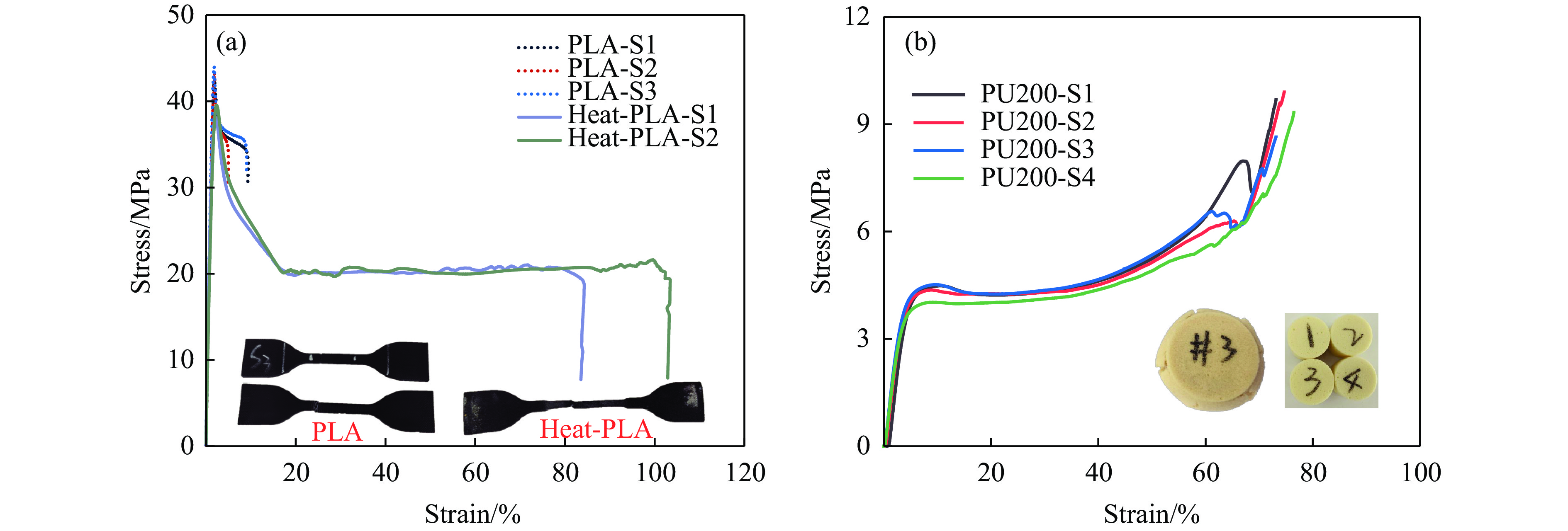
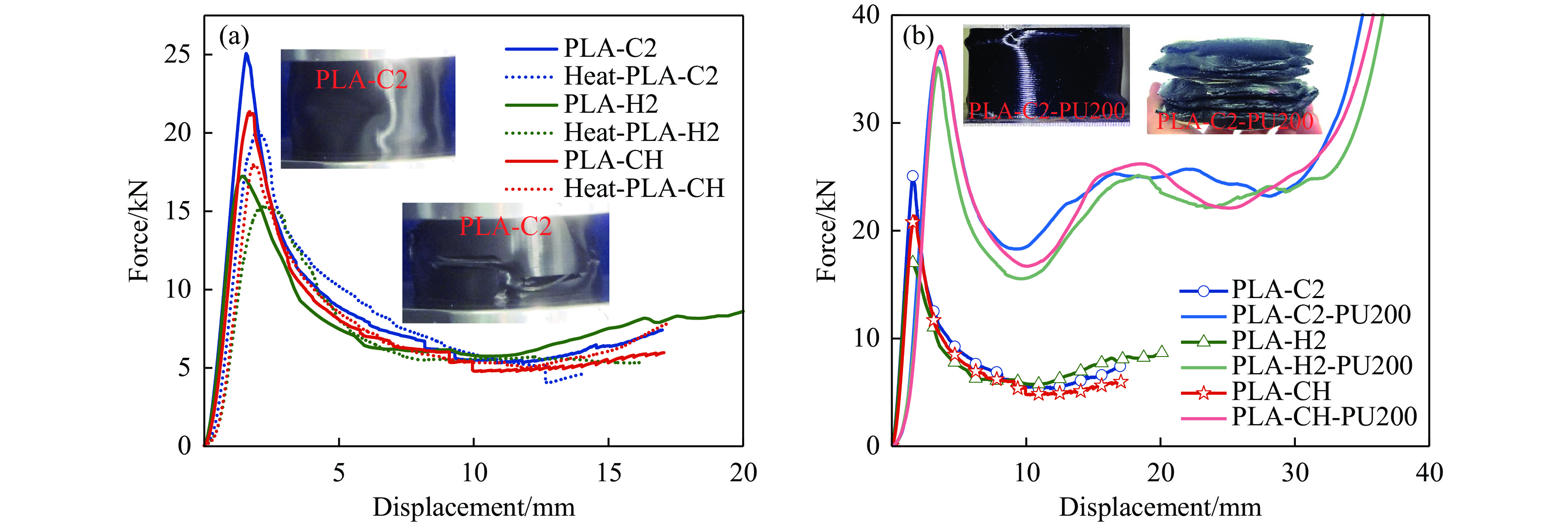
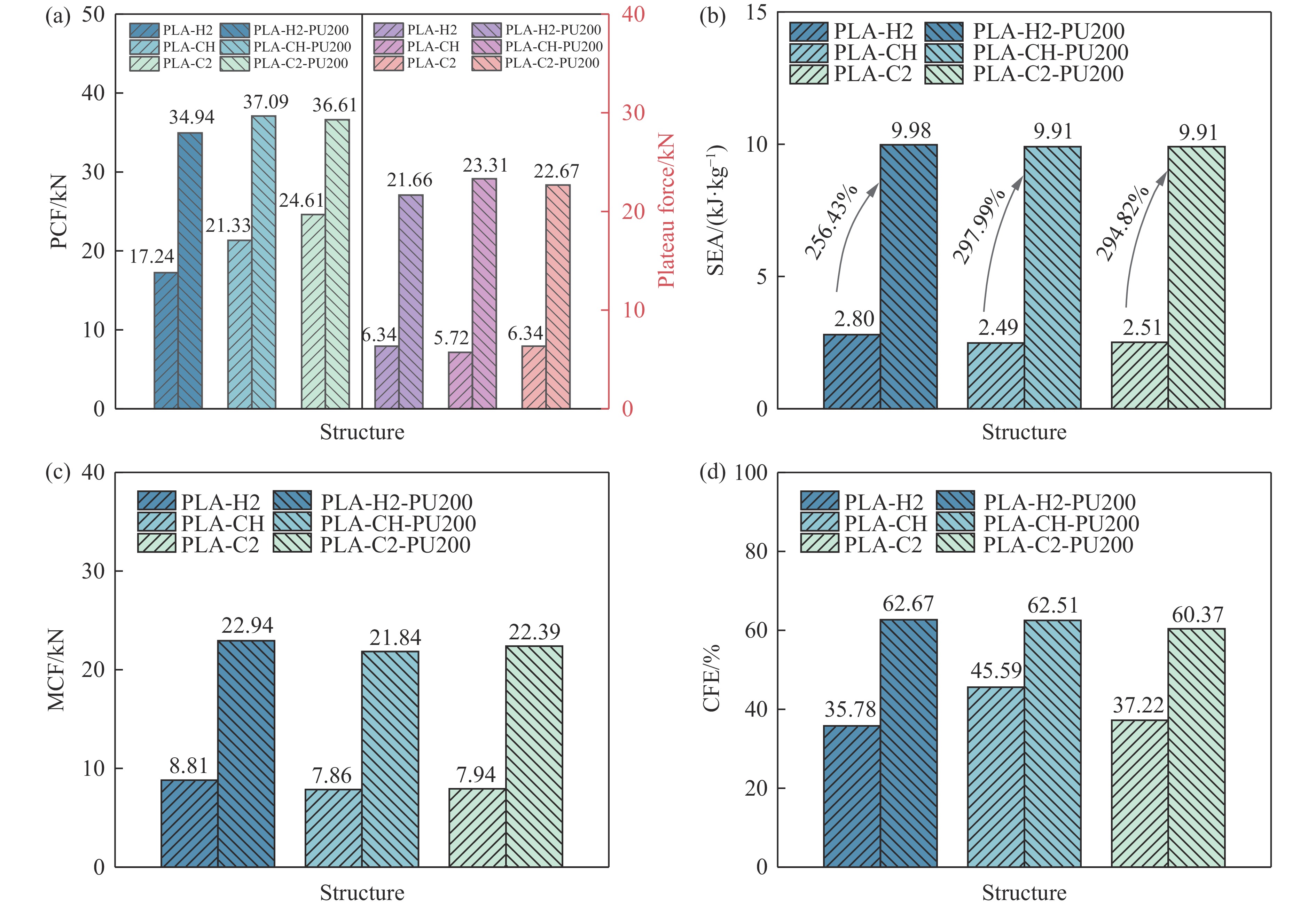
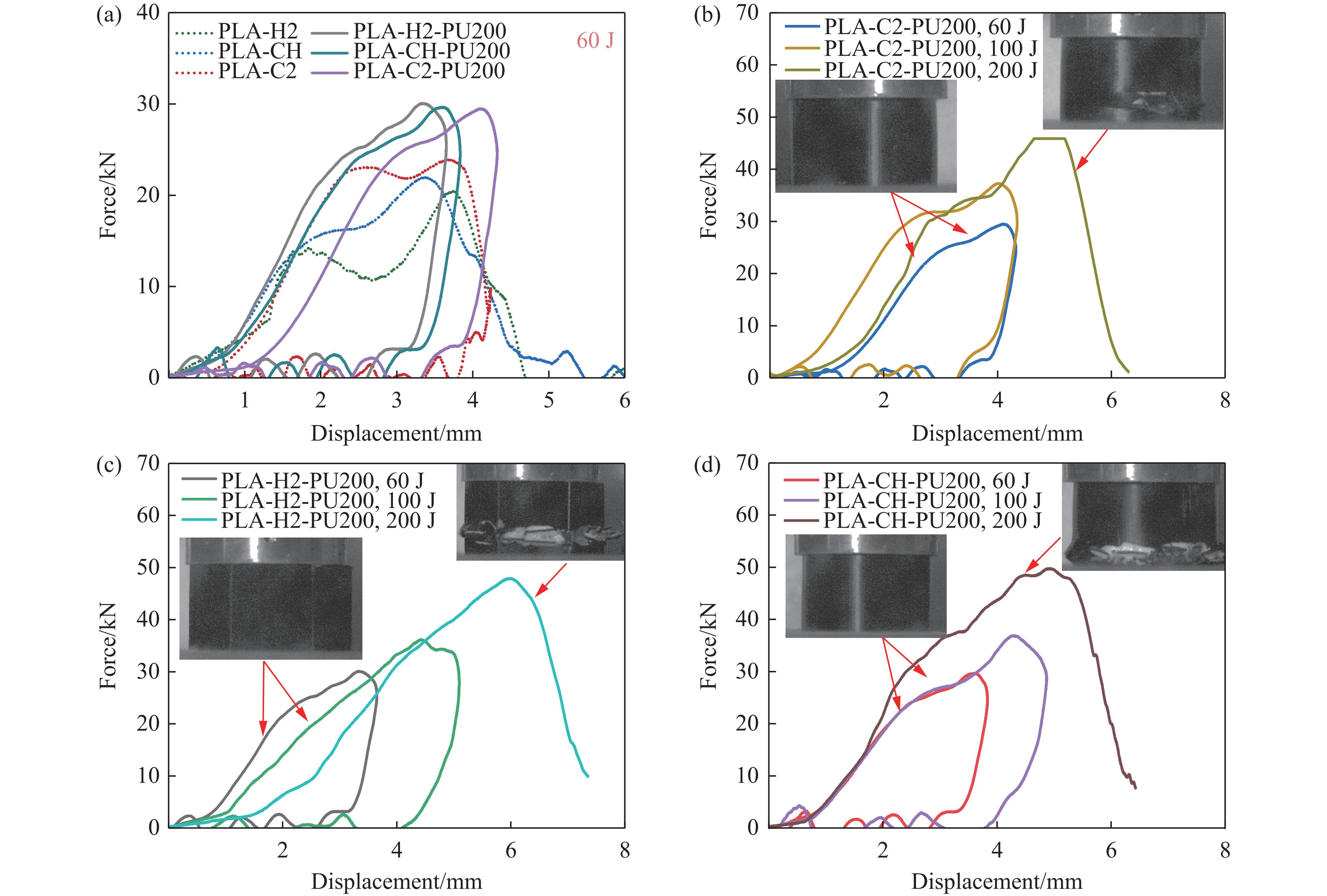
To achieve the synergistic improvement of load-bearing stability and energy absorption in lightweight protective structures, a bio-inspired thin-walled-foam composite structure based on mechanical matching design was proposed. Three configurations of polylactic acid (PLA) bio-inspired shells were fabricated via additive manufacturing, and subsequently filled with polyurethane foam through an


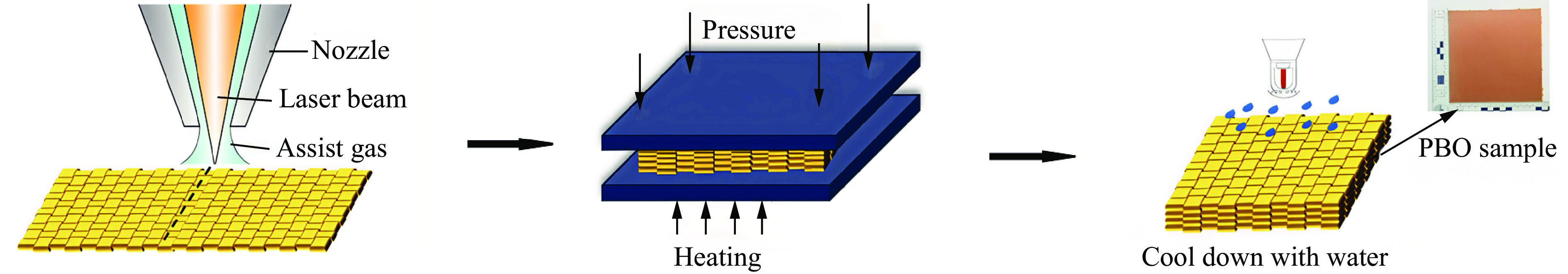
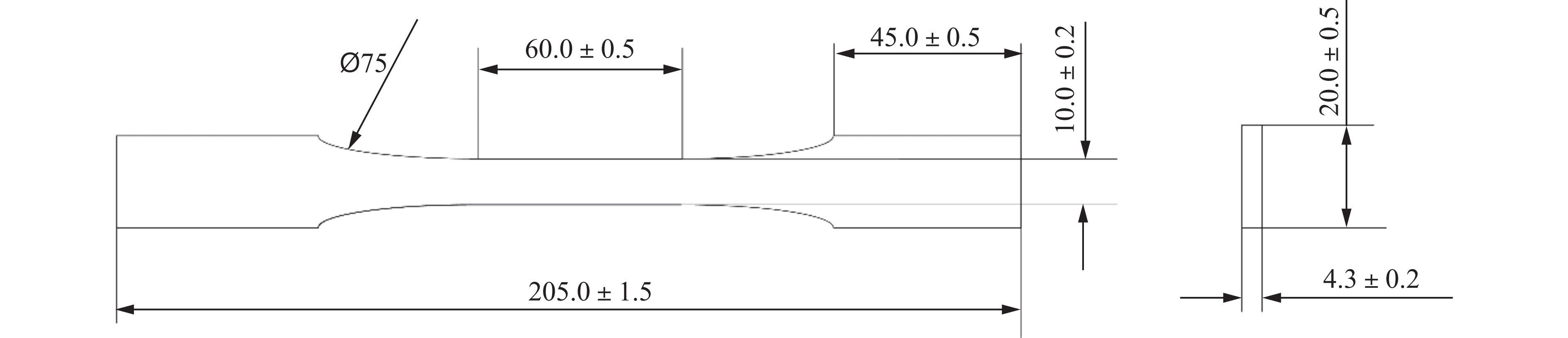
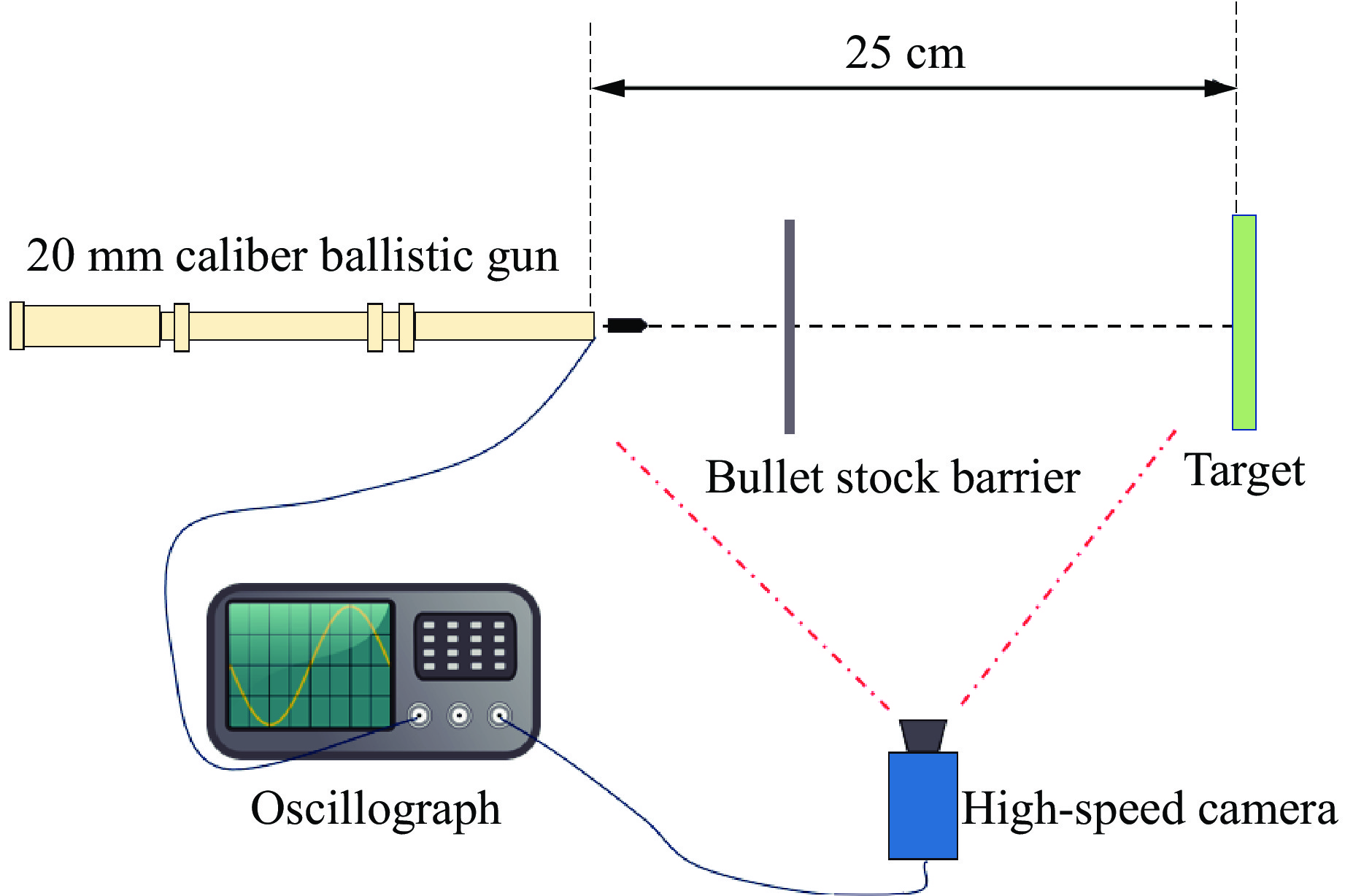
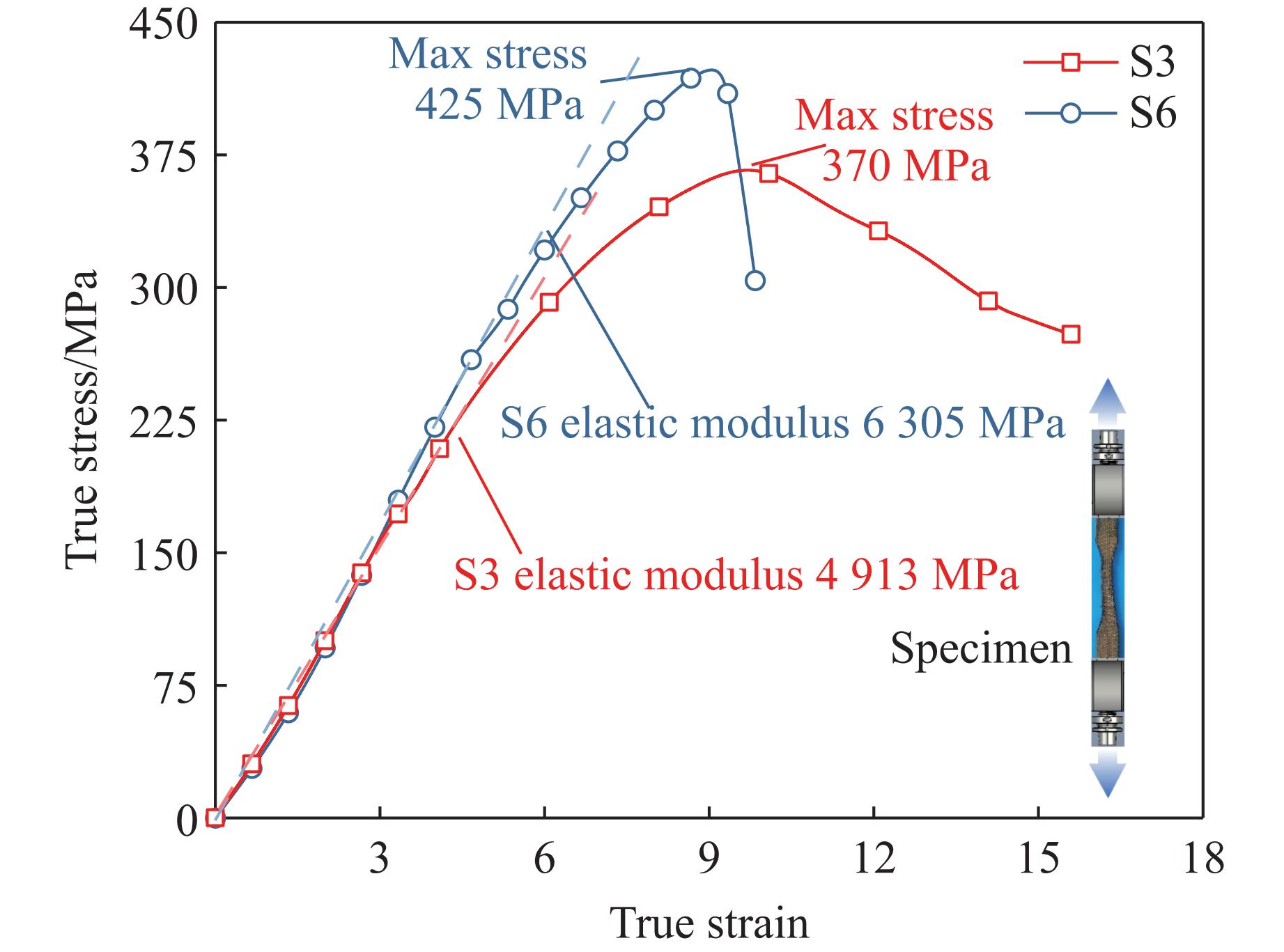
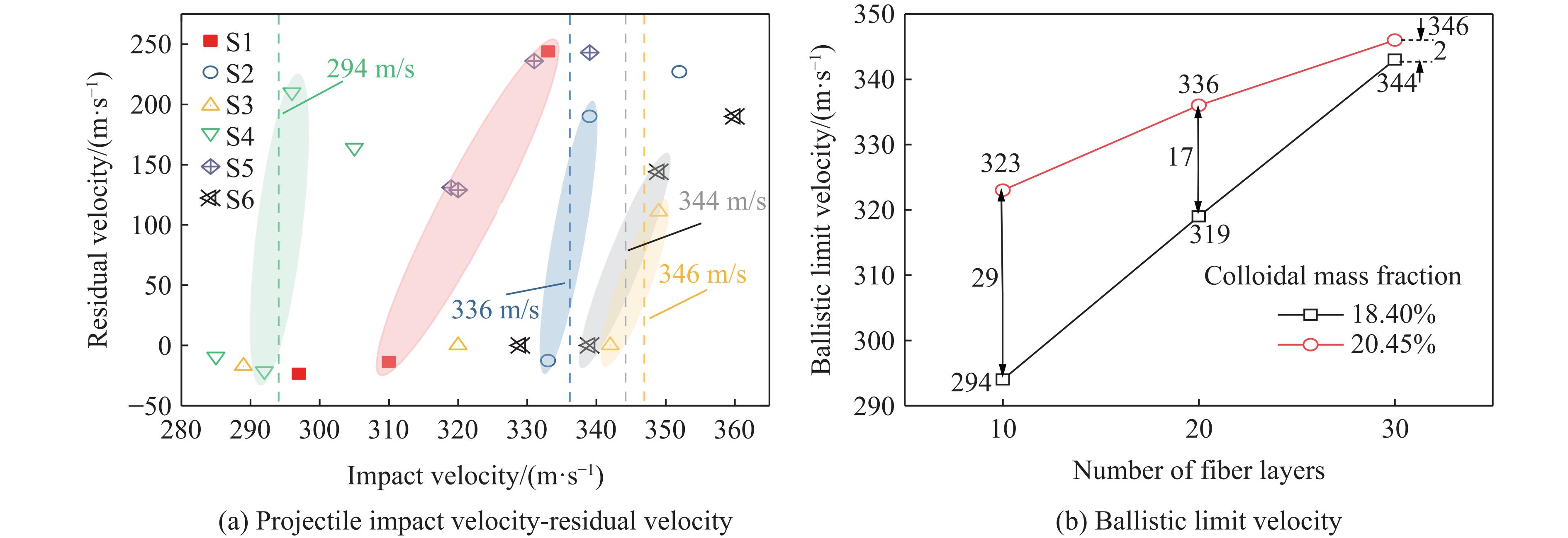
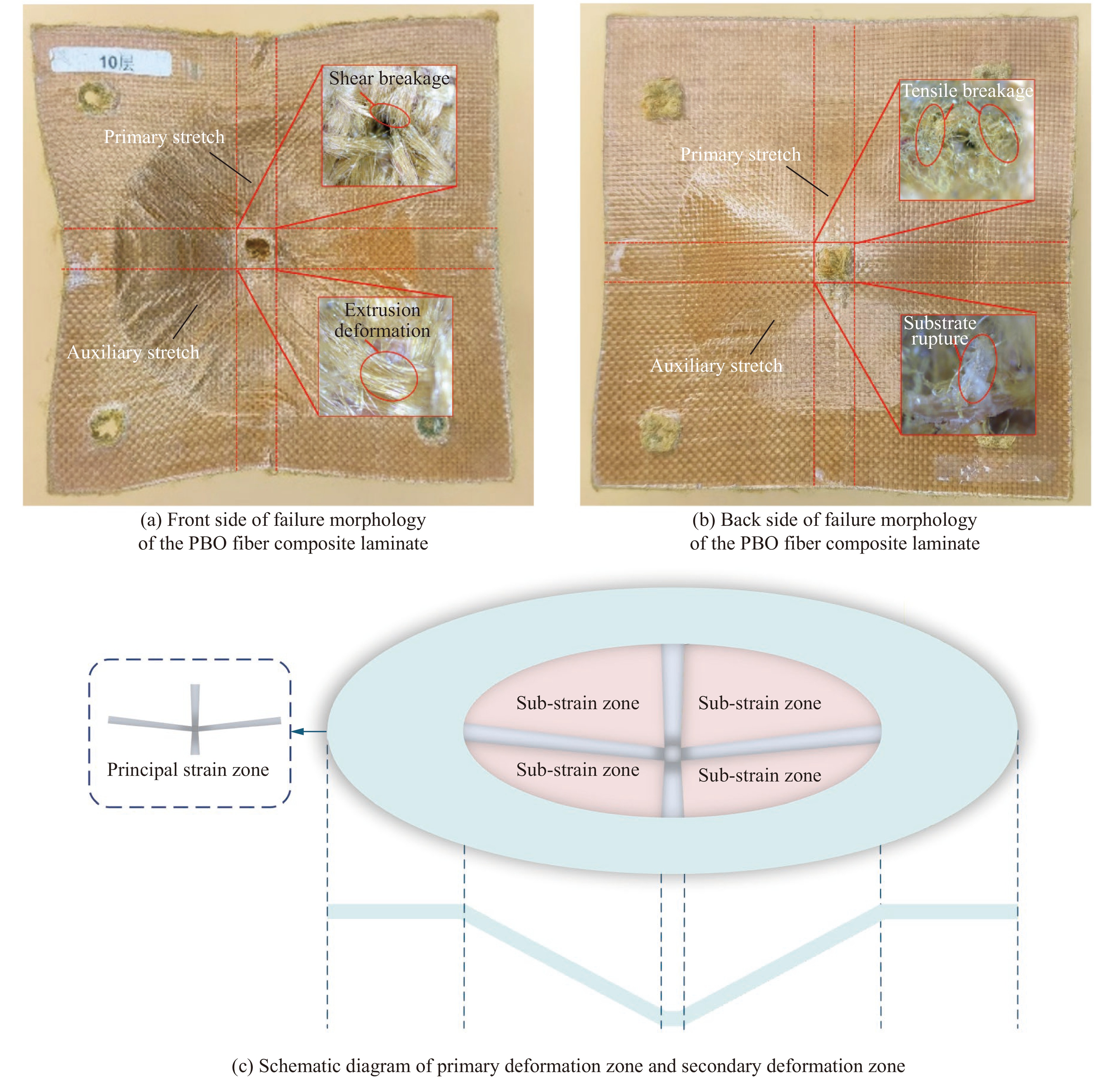
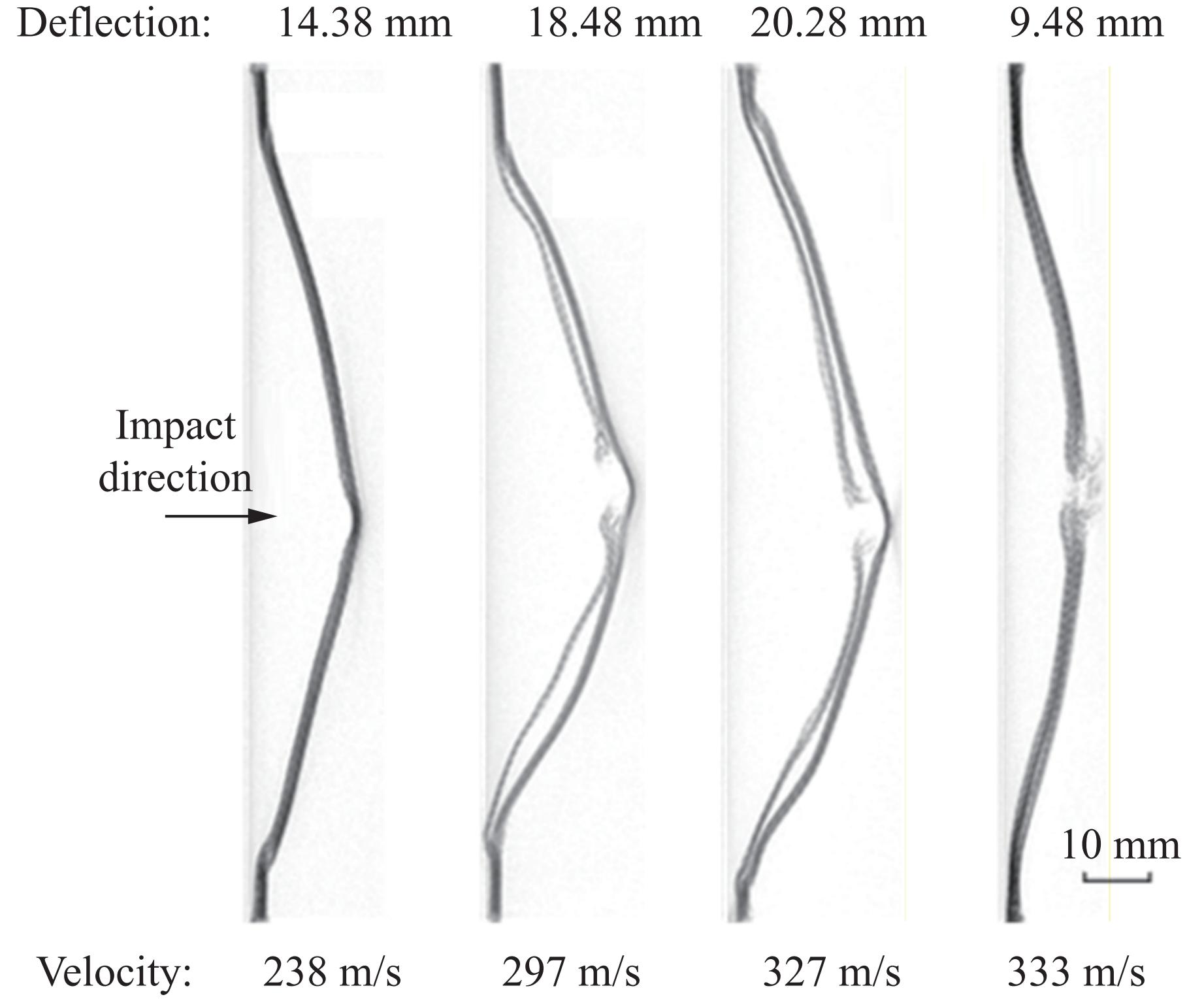
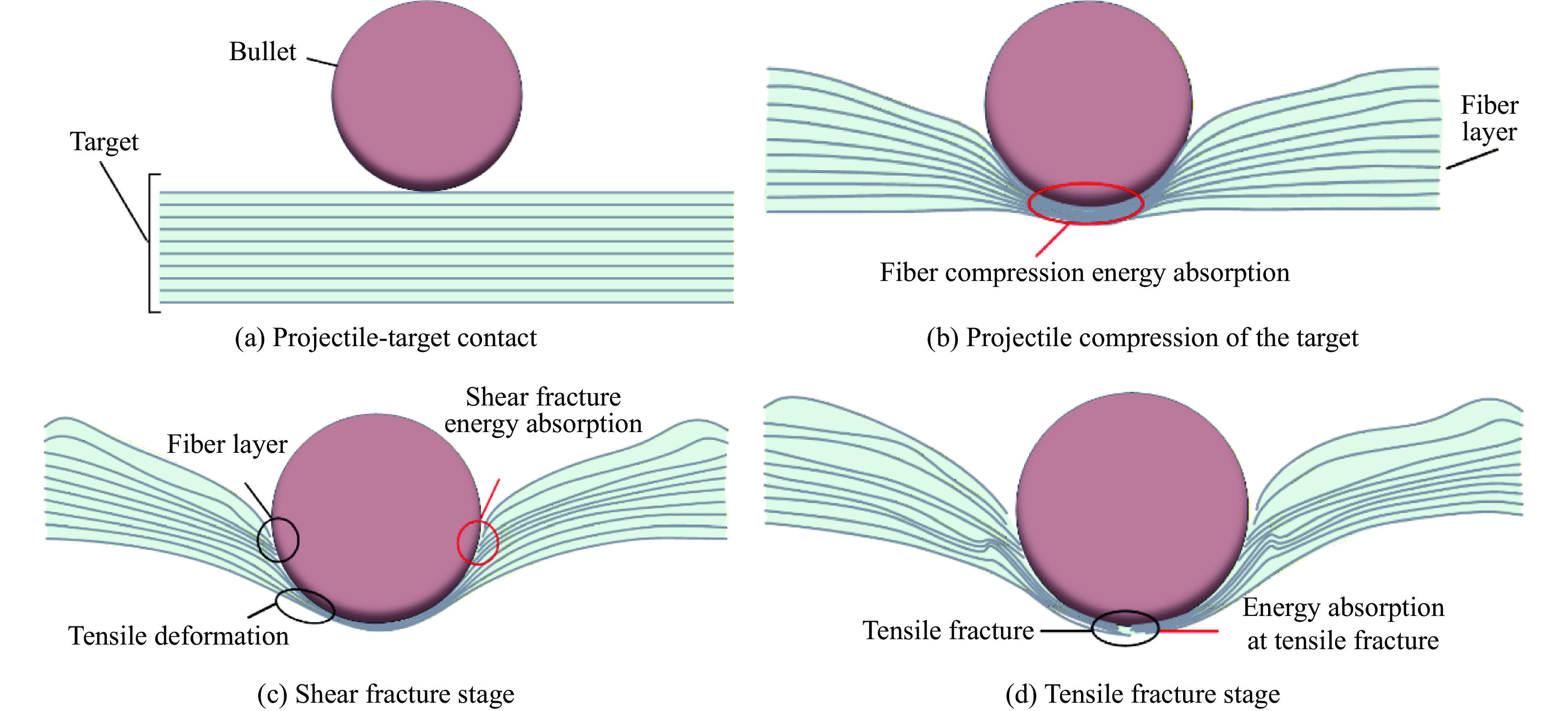
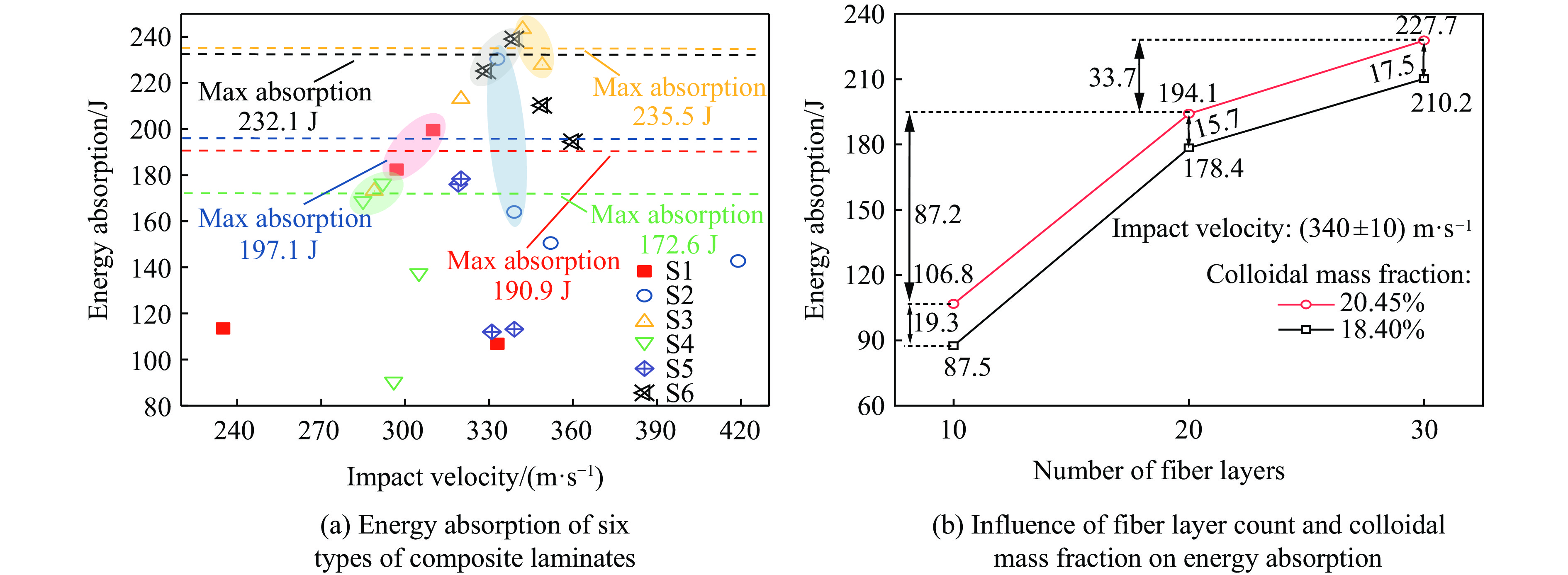
To address the issue of weak interfacial bonding and insufficient ballistic performance in poly-p-phenylene ben-zobisoxazole (PBO) fiber composites caused by the chemically inert fiber surface, the waterborne polyurethane impregnation followed by a hot-pressing process was employed to fabricate PBO composite laminates with varying resin mass fractions (18.40% and 20.45%) and numbers of fiber layers (10, 20, and 30 layers). The tensile properties, ballistic limit, failure modes, and energy absorption mechanisms of the composites were investigated through quasi-static tensile testing, ballistic experiments, and X-ray computed tomography (CT). The results indicate that this process can effectively improve the formability and protective performance of the PBO fiber composite laminates. In comparison with the specimen exhibiting a resin mass fraction of 20.45%, the 30-layer specimen with a resin mass fraction of 18.40% demonstrated a 14.86% increase in tensile strength and a 28.33% increase in elastic modulus. The ballistic limit increases with resin content. The ballistic limit of the 10-layer and 20-layer specimens with a resin mass fraction of 20.45% increases by 9.9% and 5.3%, respectively. However, the increasing effect diminishes with a greater number of layers. For the 30-layer specimens, the difference in resin mass fraction between the two resins was less than 1%. The primary failure modes of the composite laminates include fiber shear fracture, matrix cracking and delamination, and fiber tensile fracture. Energy absorption is achieved through the synergistic mechanisms of fiber compressive deformation, shear, and tensile fracture. Efficiency of the energy absorption decreases with increasing impact velocity, while it increases with the number of layers under the same impact velocity. The effect of colloid content diminishes significantly for numerous layers. The study can provide a reference for the design of ballistic protection using PBO fiber composites.


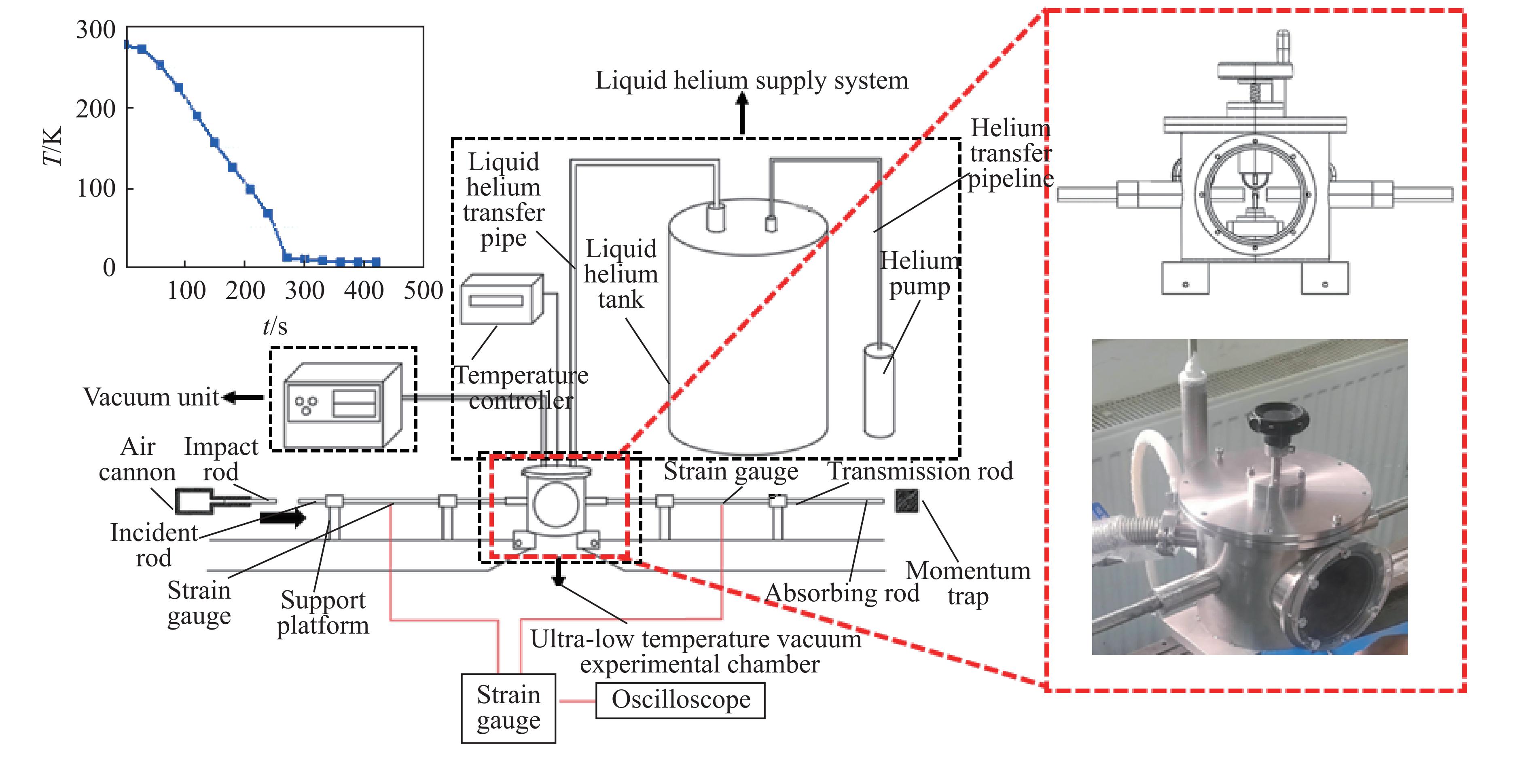
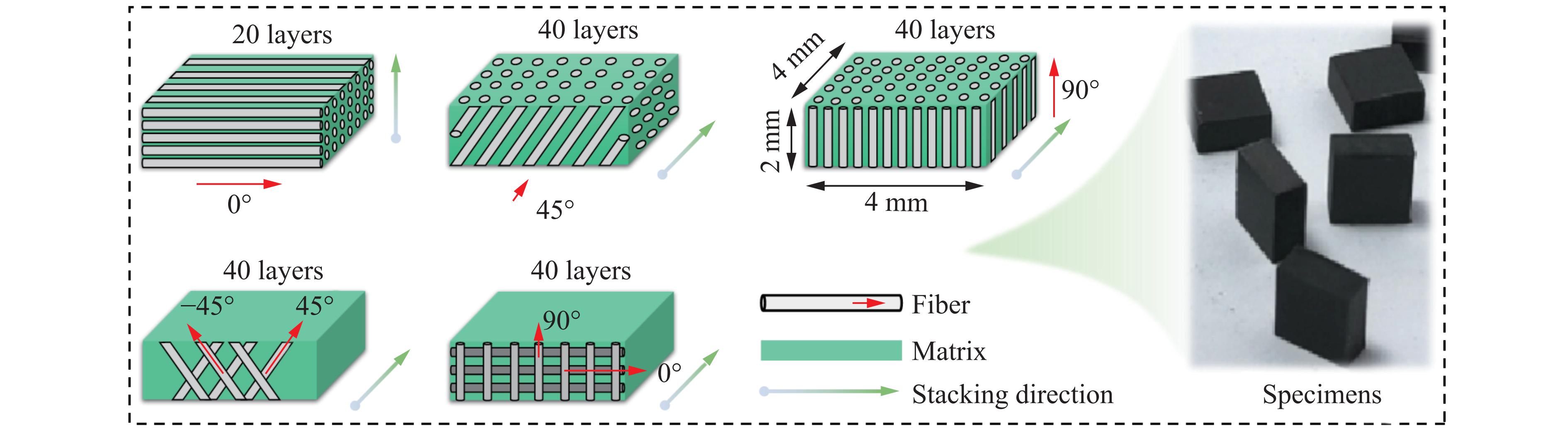
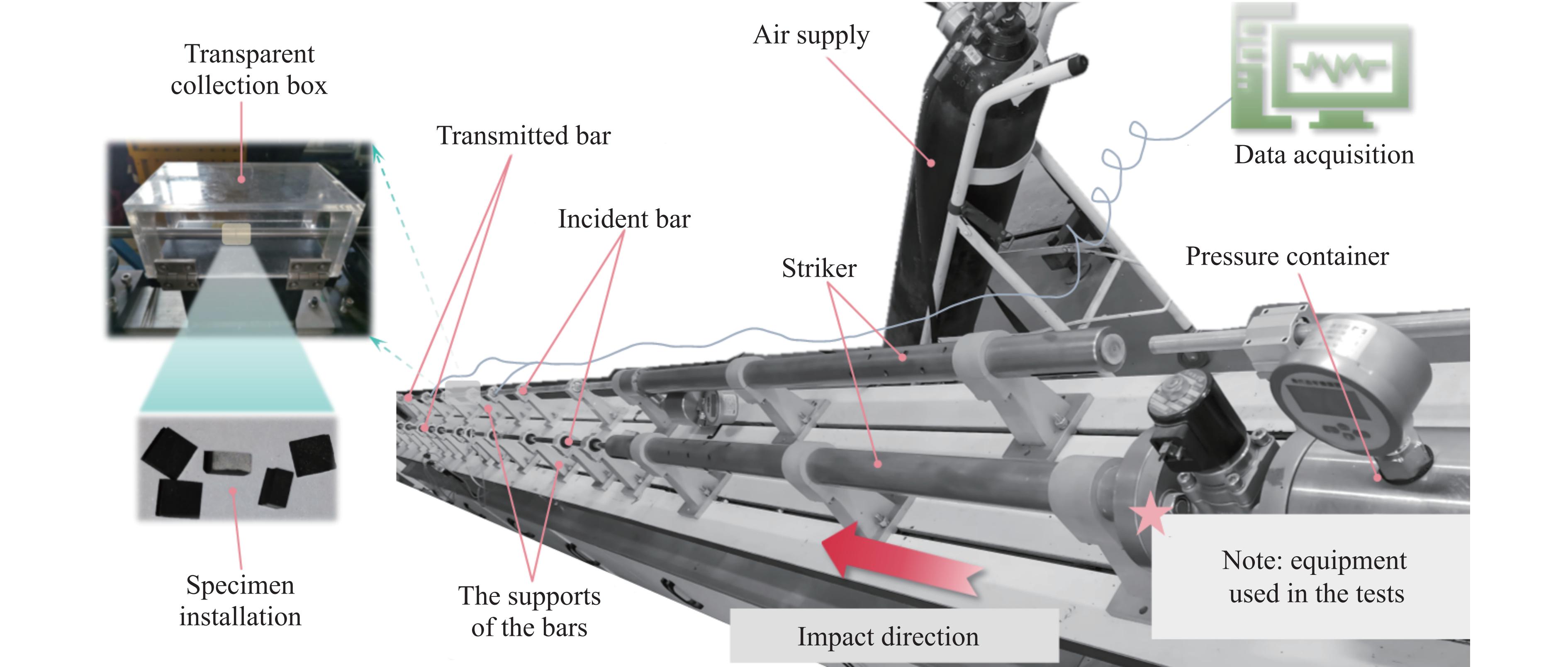
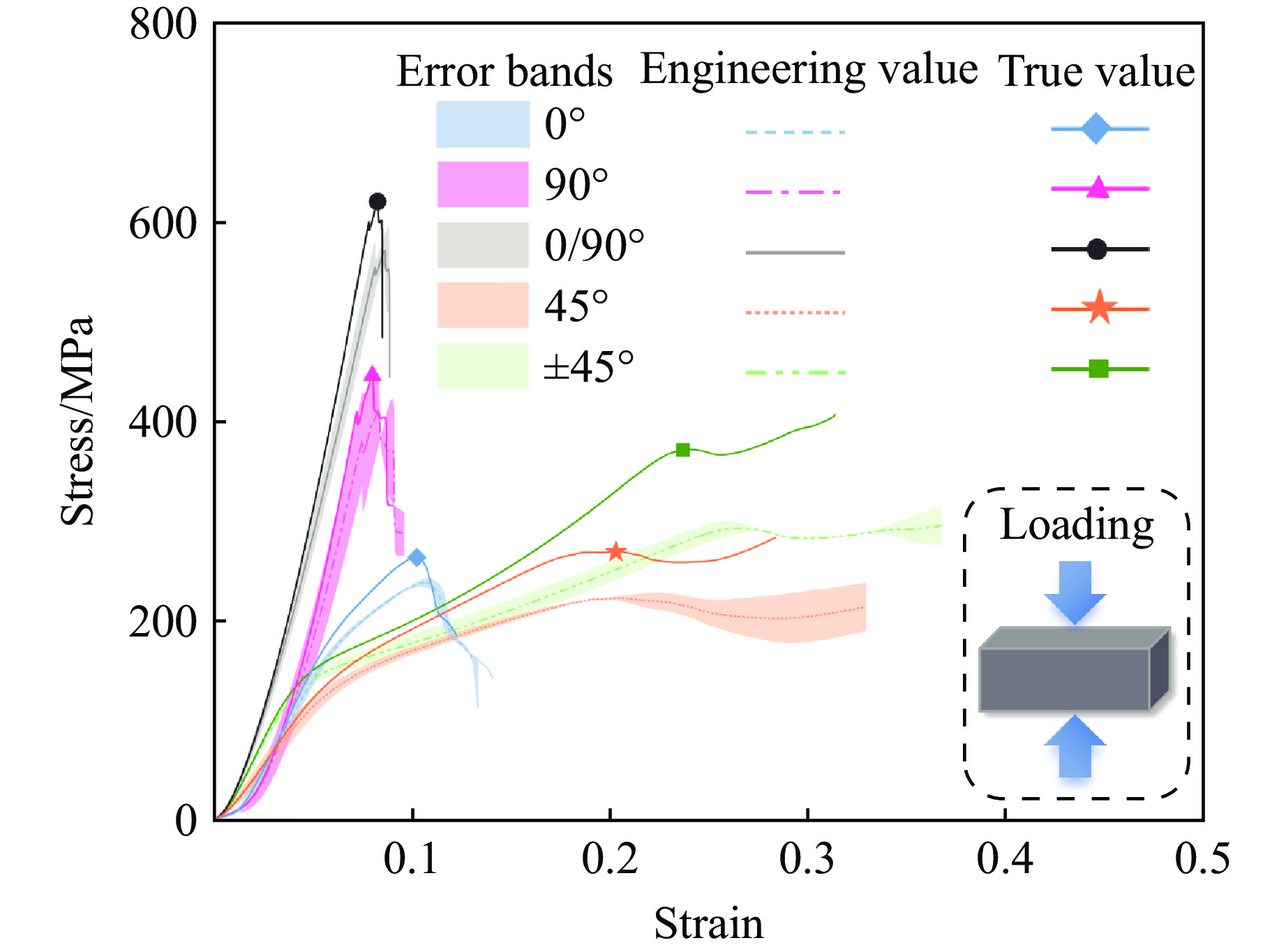
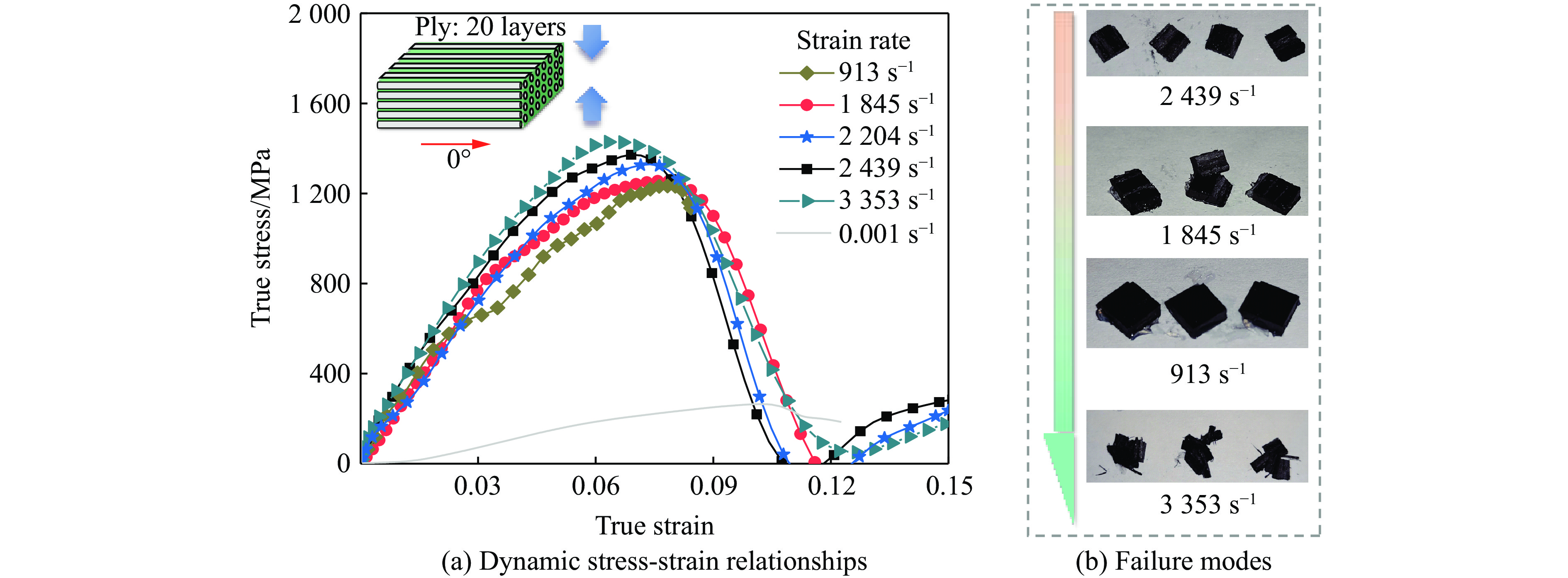
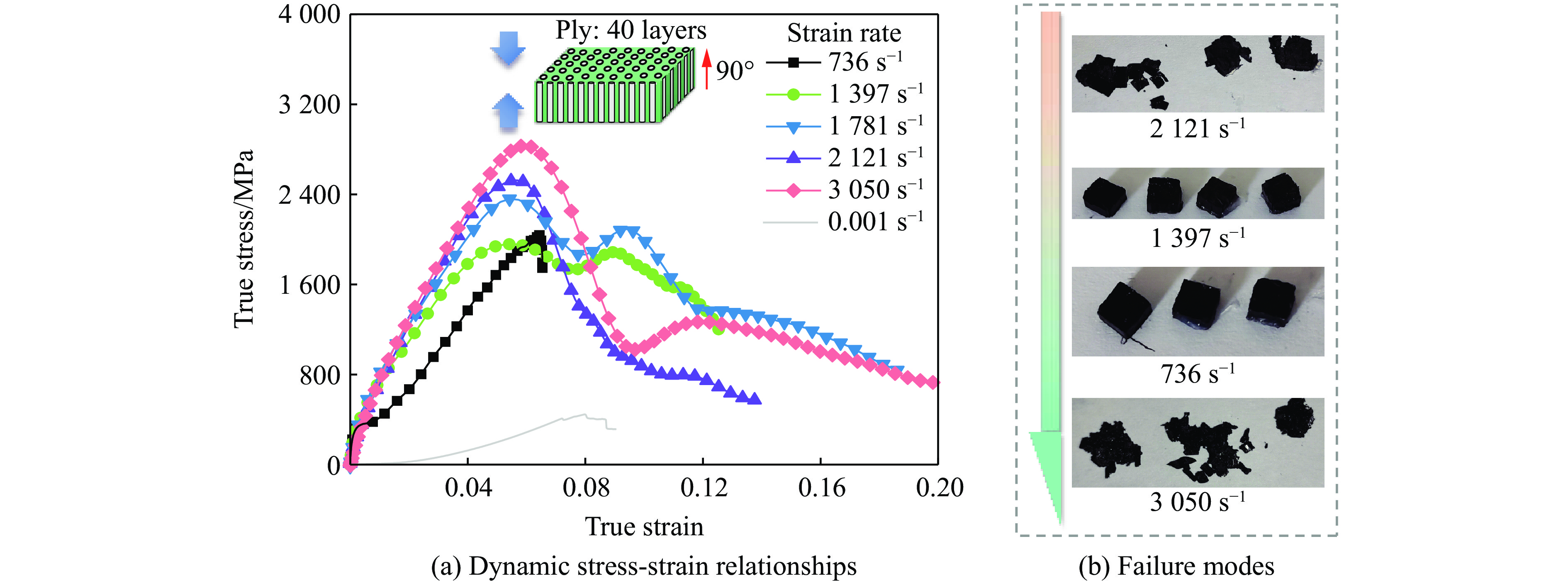
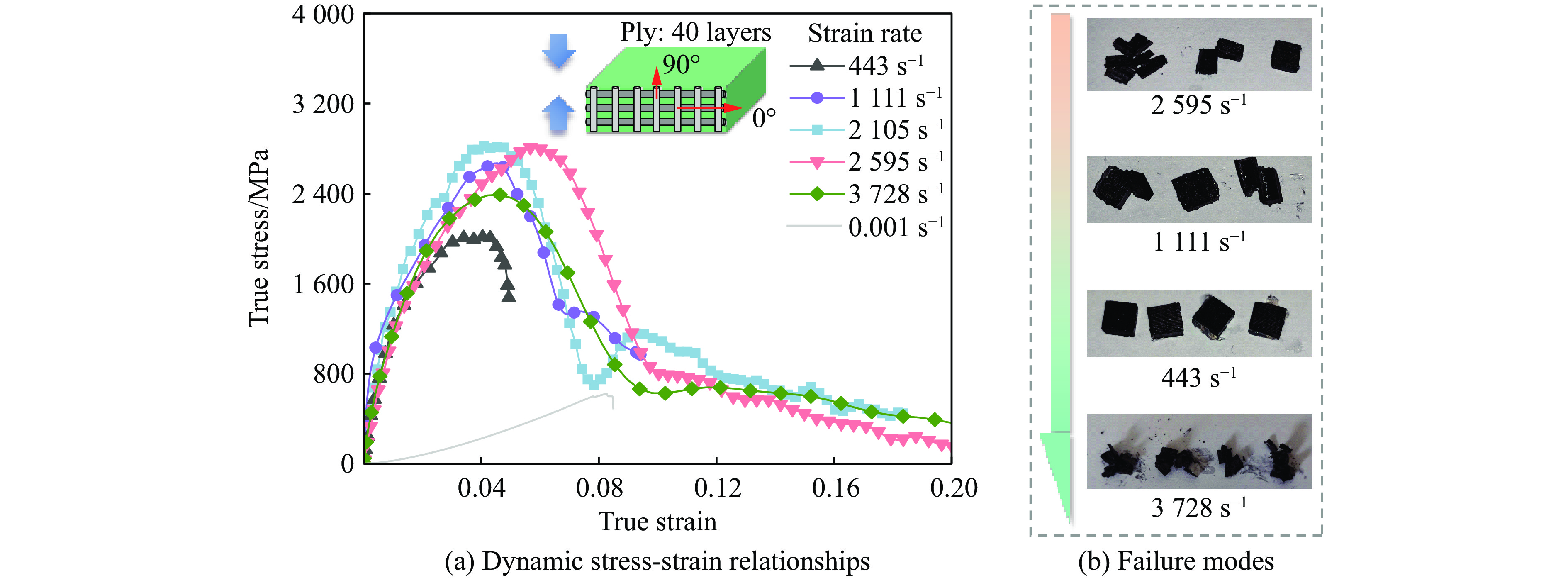
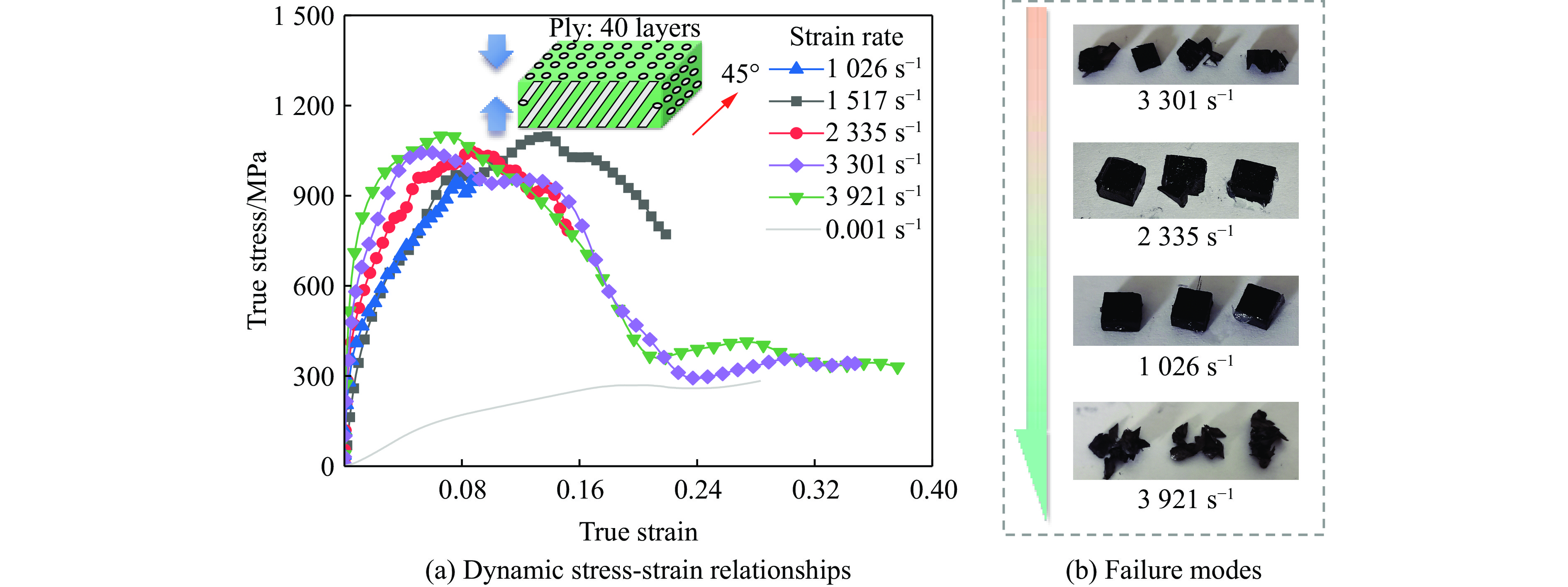
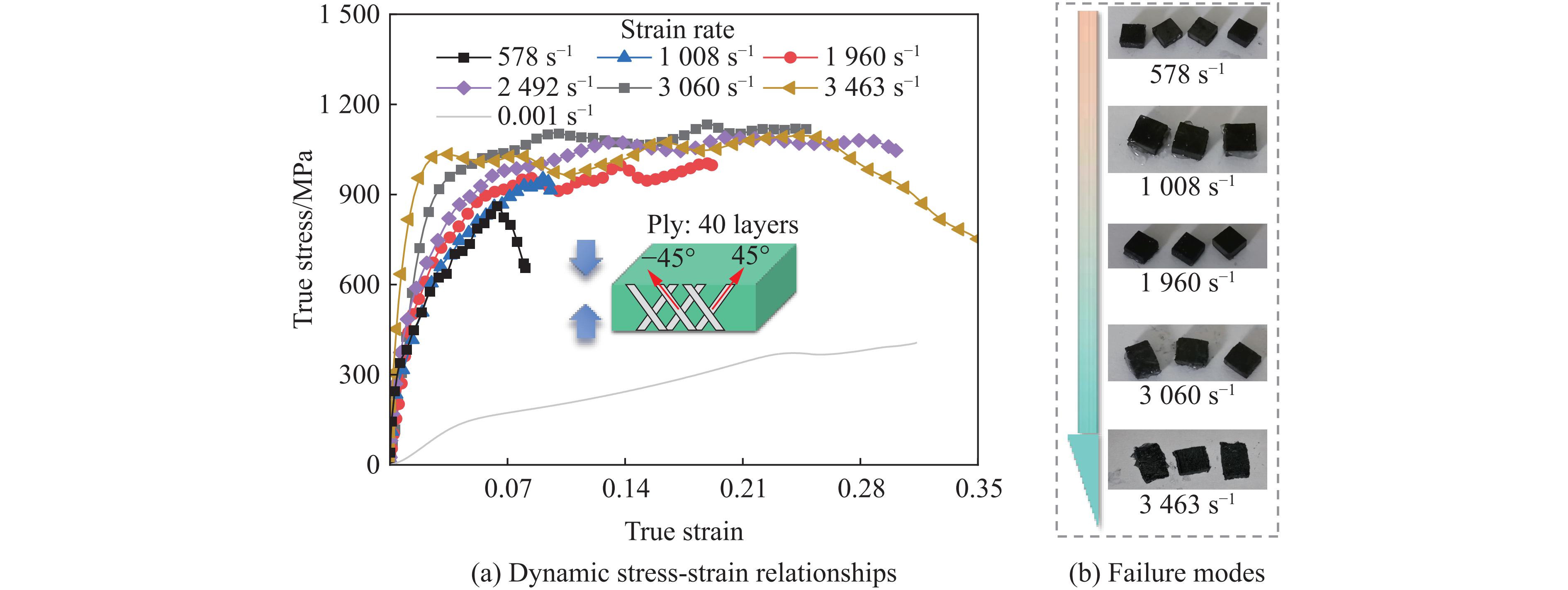
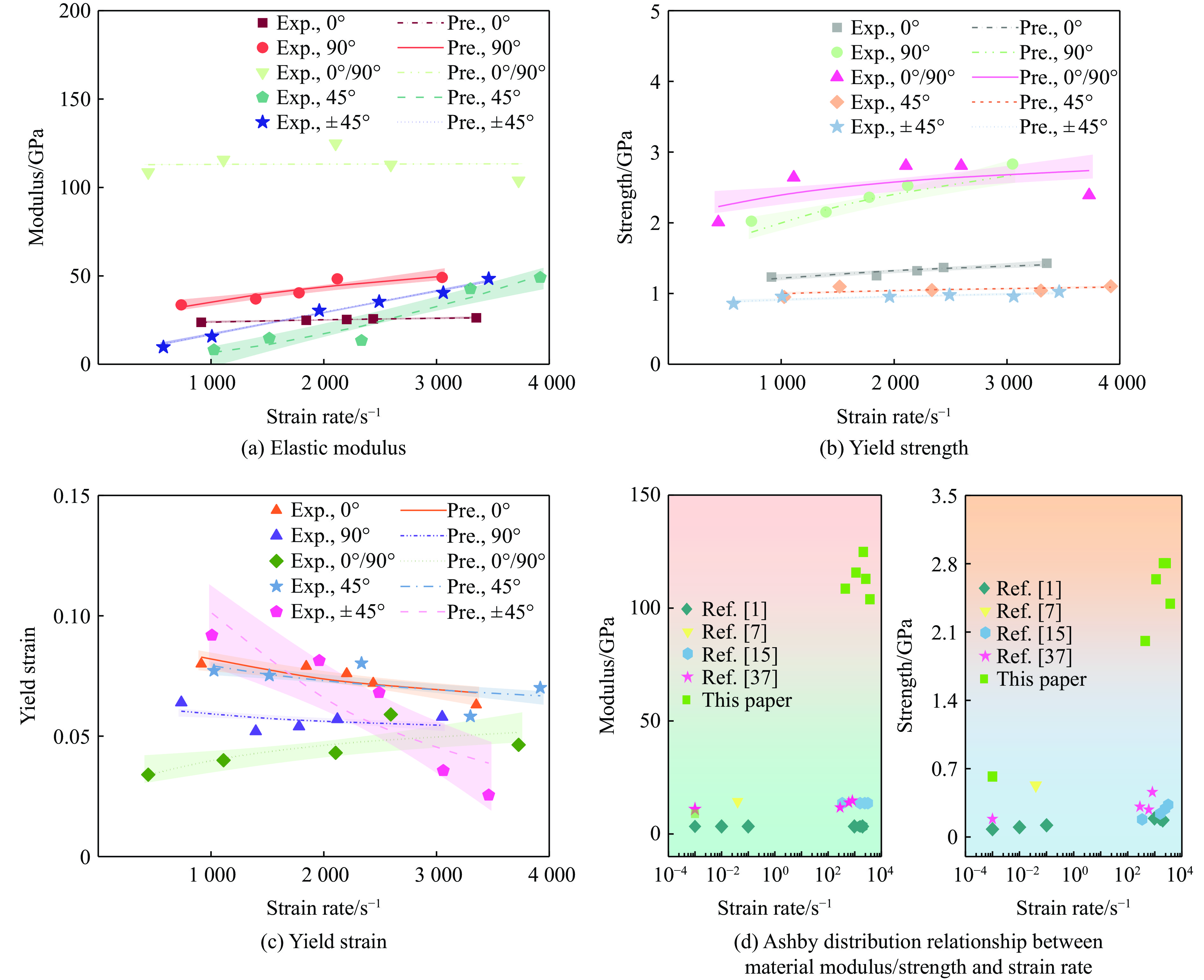
Carbon fiber reinforced polymer (CFRP), as an advanced composite material, are widely used in engineering applications. However, research on the dynamic mechanical behavior of ultrathin CFRP laminates remains relatively limited. In this study, unidirectional ultrathin prepreg and hot-pressing molding processes were employed to fabricate ultrathin CFRP laminates with a single ply thickness of only 0.1 mm. The strain rate effects on specimens with five different ply orientations—0°, 90°, 0°/90°, 45°, and ±45°—were systematically investigated. Quasi-static compression experiments indicated that the 45° ply orientation enhanced plastic behavior but reduced material strength and modulus, whereas the 90° ply orientation contributed to increased modulus and strength while reducing plastic deformation. Dynamic impact tests revealed that the 90° ply orientation improved both dynamic modulus and strength while decreasing yield strain. Although the 45° ply orientation reduced dynamic yield strength, it significantly increased the sensitivity of dynamic modulus and yield strain to strain rate. Compared with conventional CFRP laminates with a 0°/90° ply layup (ply thickness is 0.295 mm, and dynamic strength and modulus are 900 MPa and 10.12 GPa, respectively), the ultrathin CFRP composites developed in this study exhibited a 66% increase in fiber content per unit thickness; under the 0°/90° ply configuration, dynamic strength and modulus were enhanced by 123% and 926%, respectively. Based on the experimental data, a constitutive model for the ultrathin CFRP composites was established, and corresponding constitutive parameters were provided, offering a basis for predicting the mechanical behavior of CFRPs under different ply orientations and strain rates.
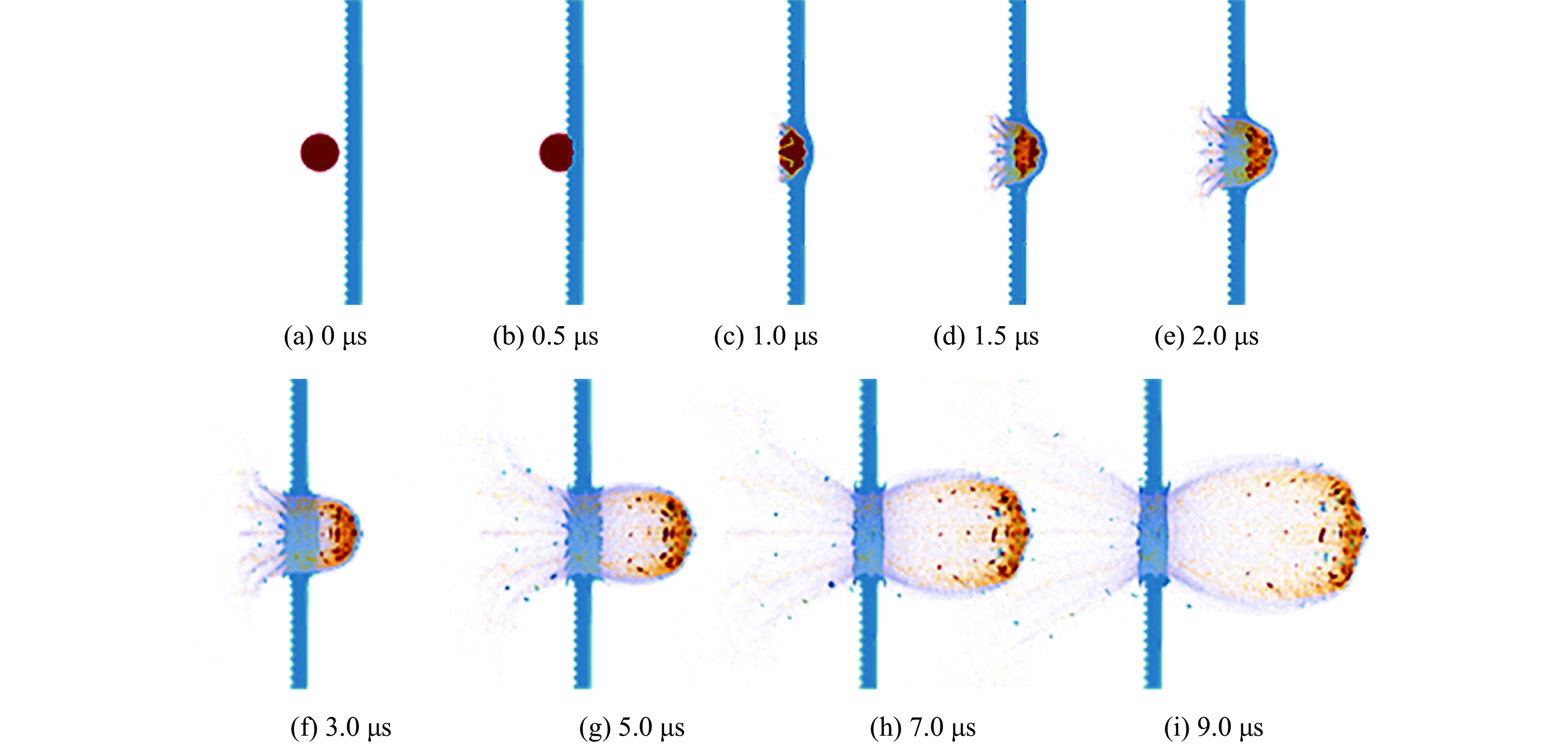
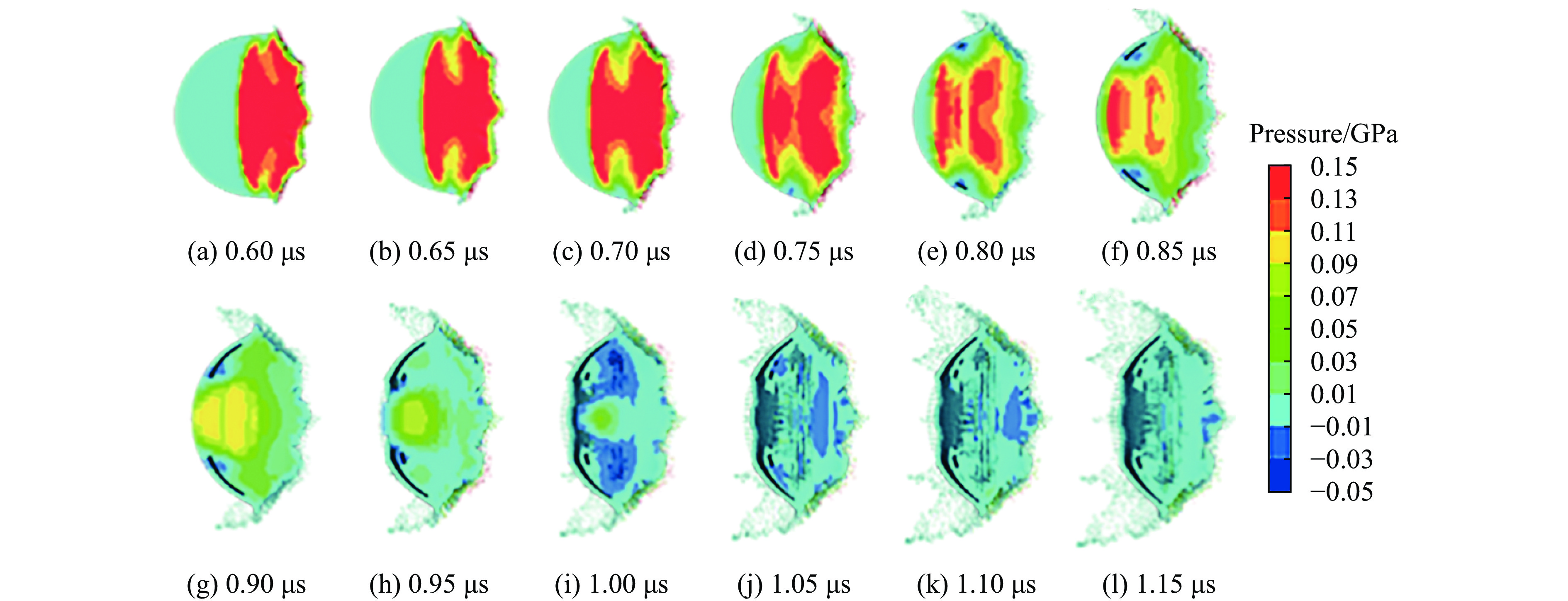
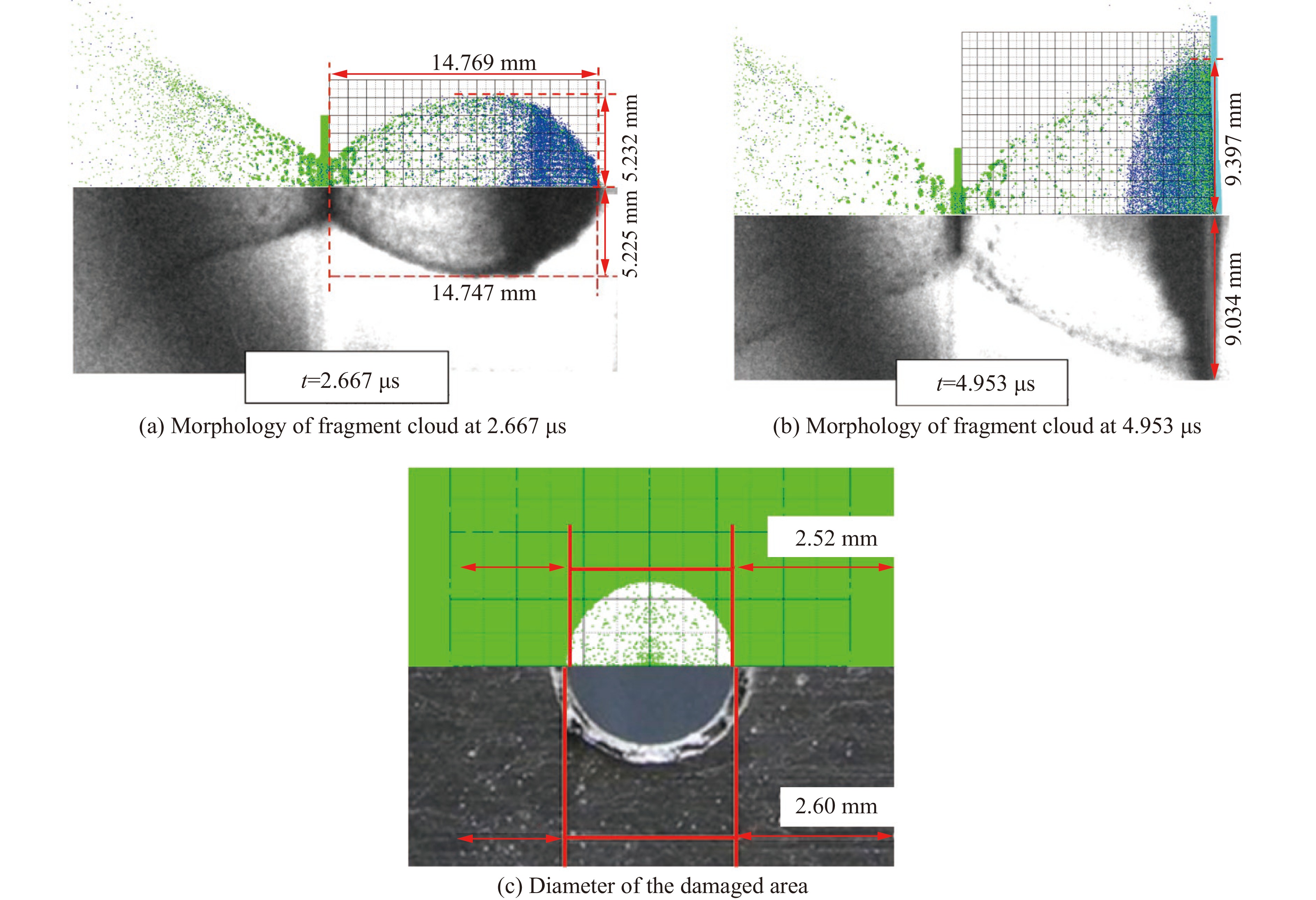
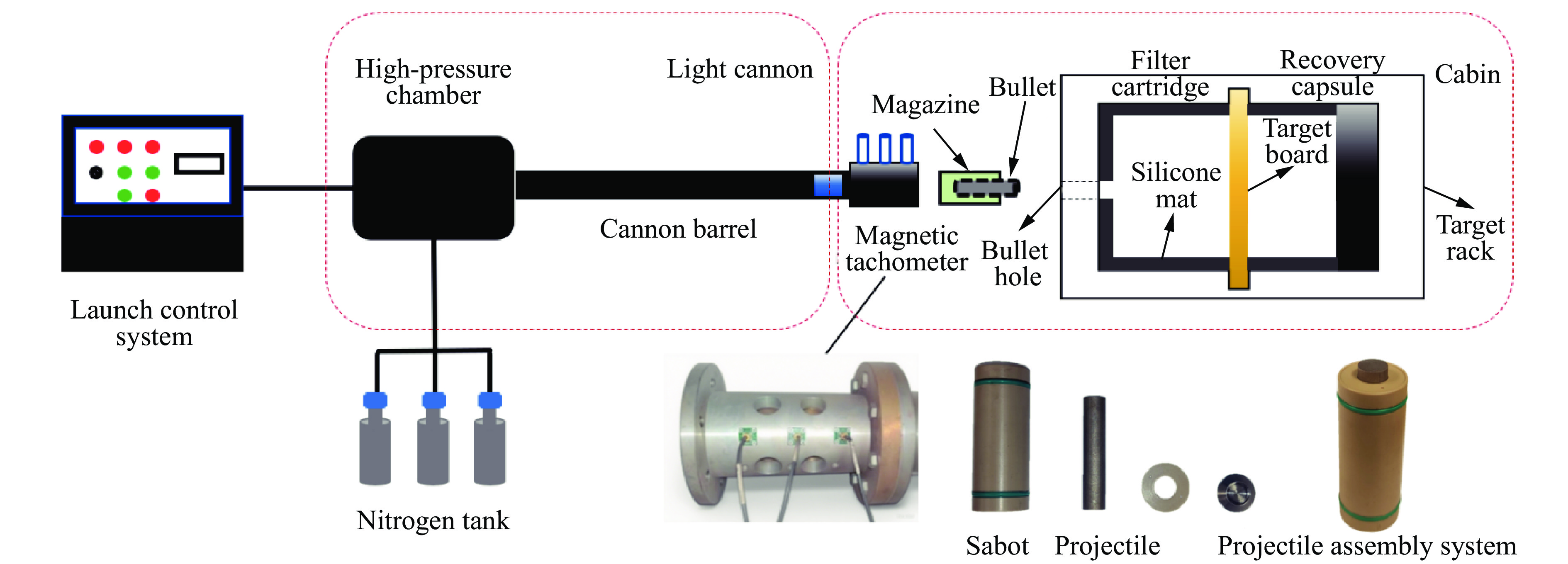
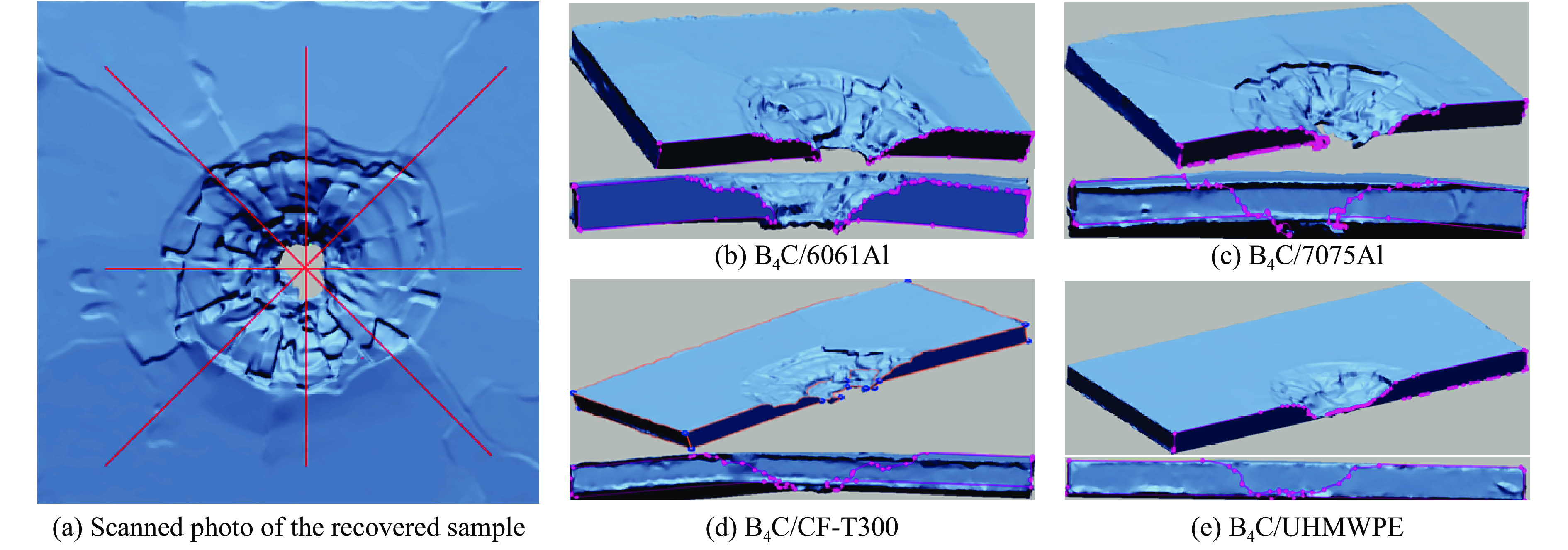
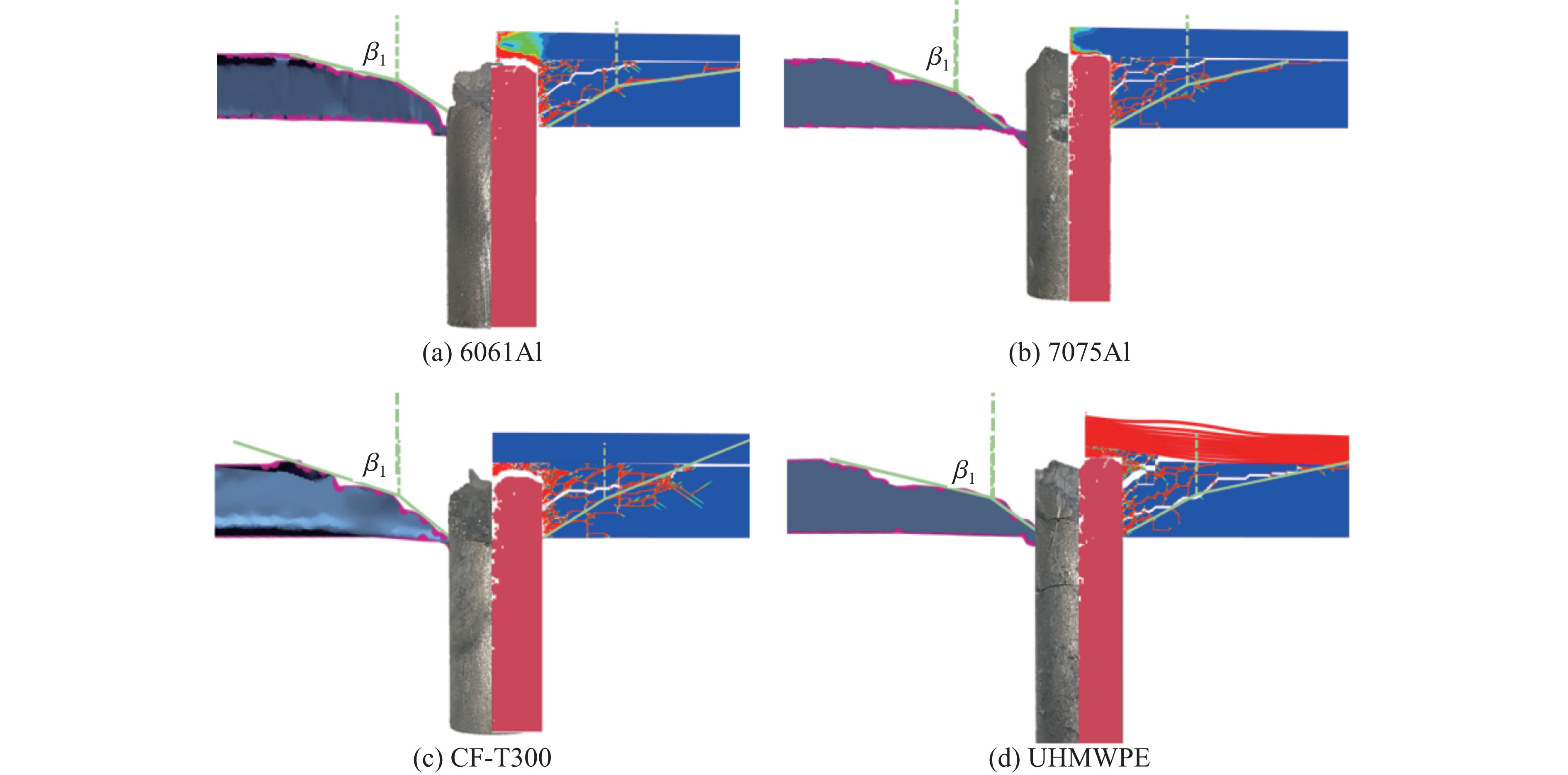
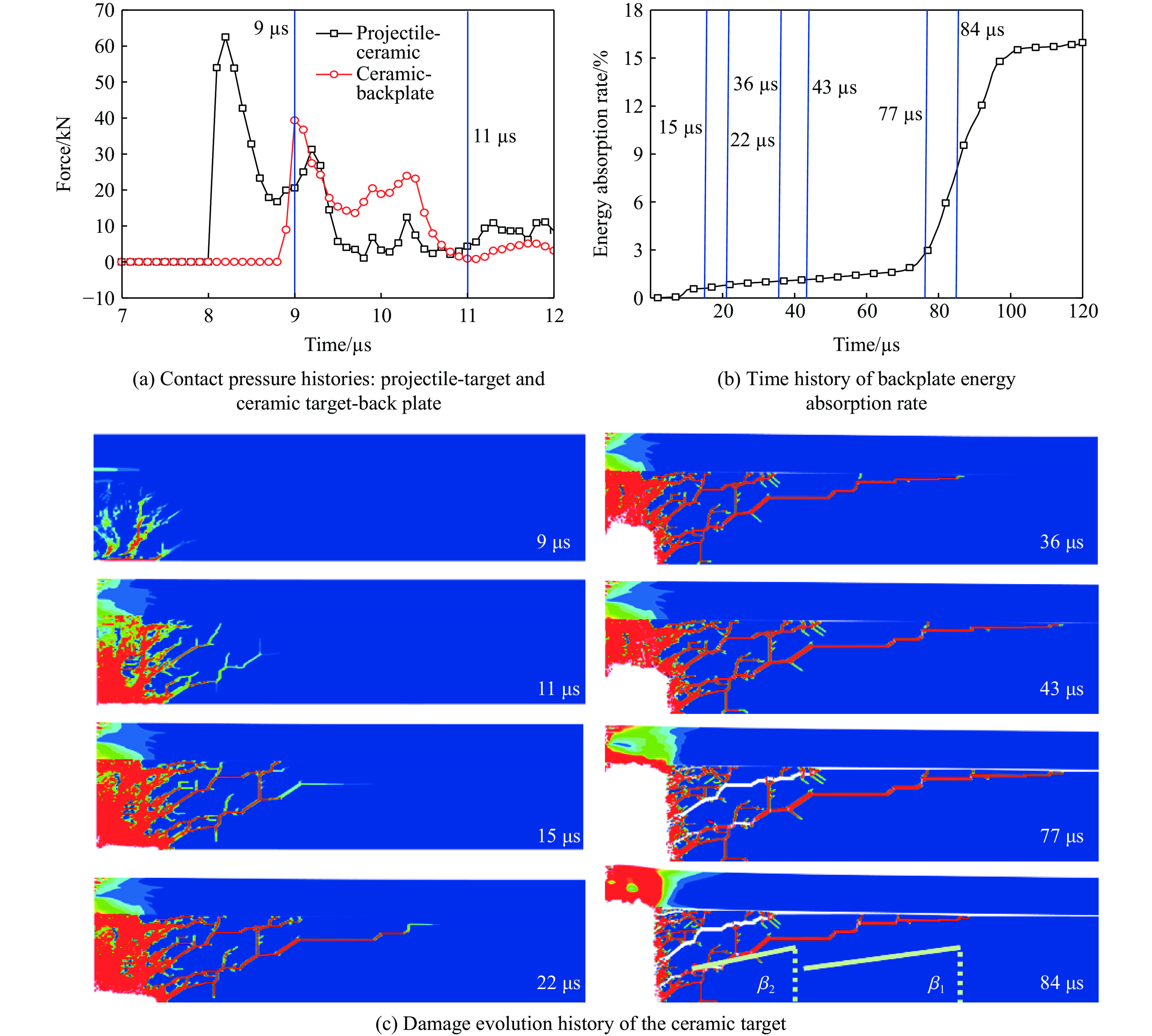
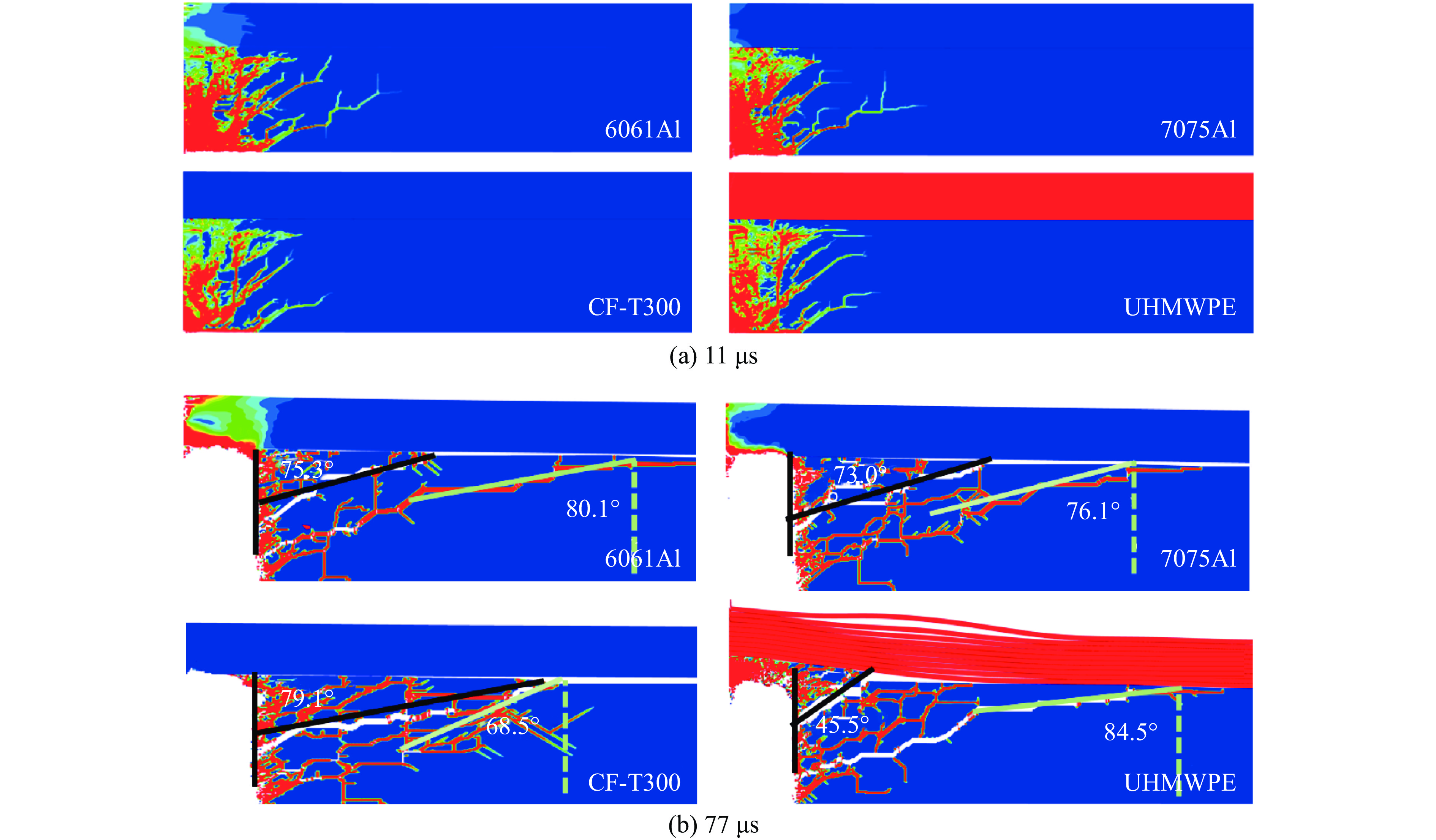
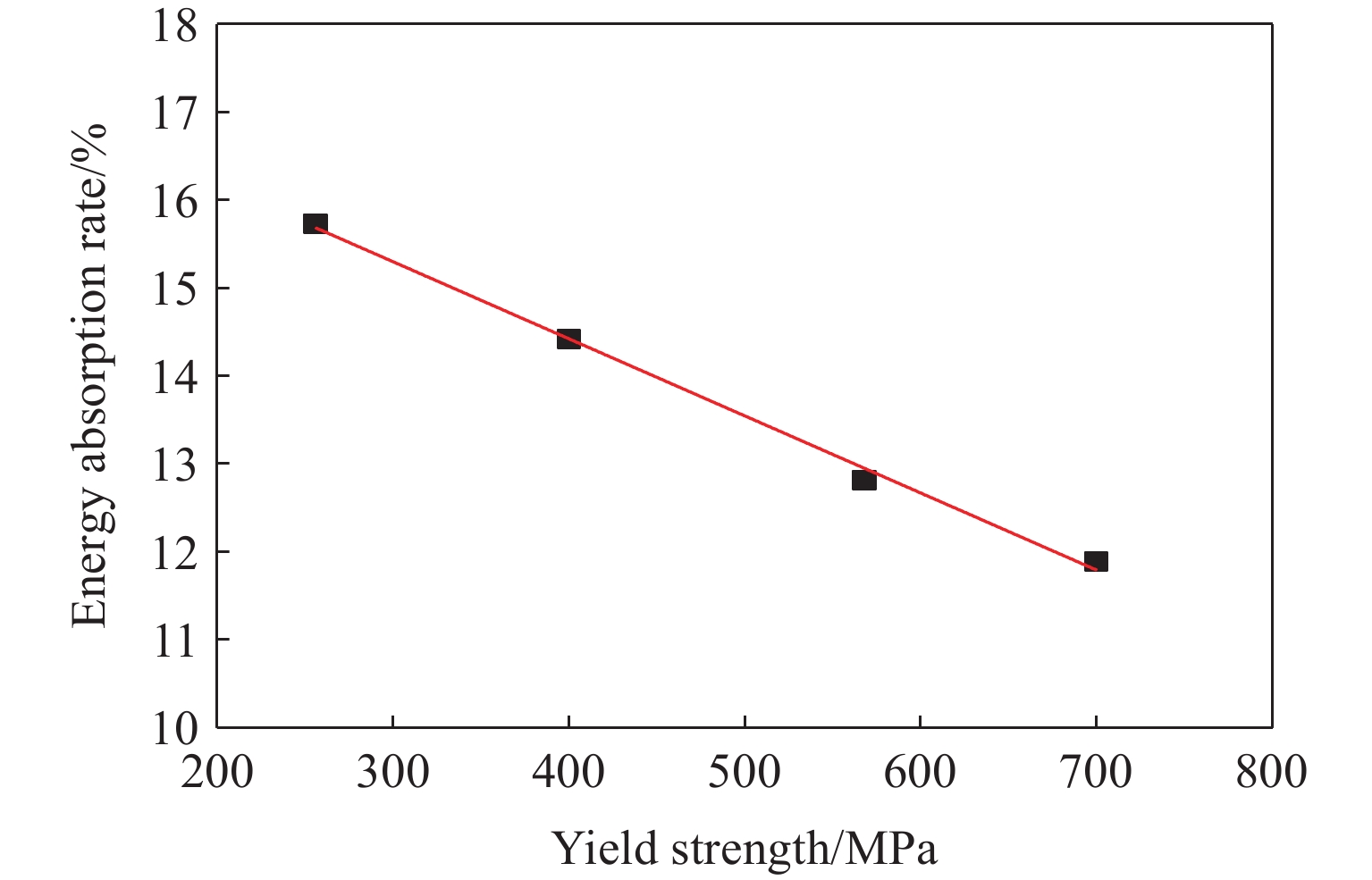
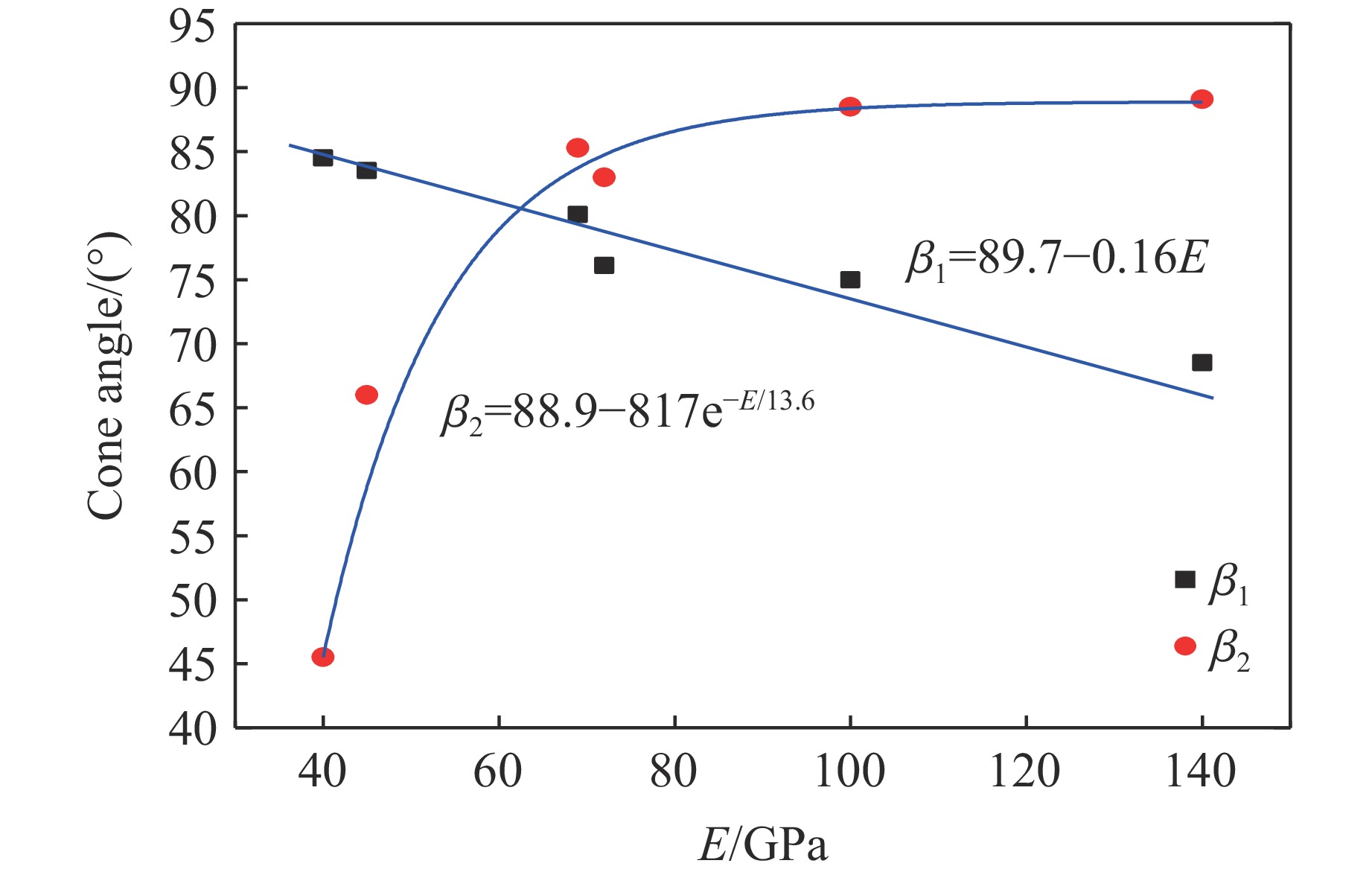
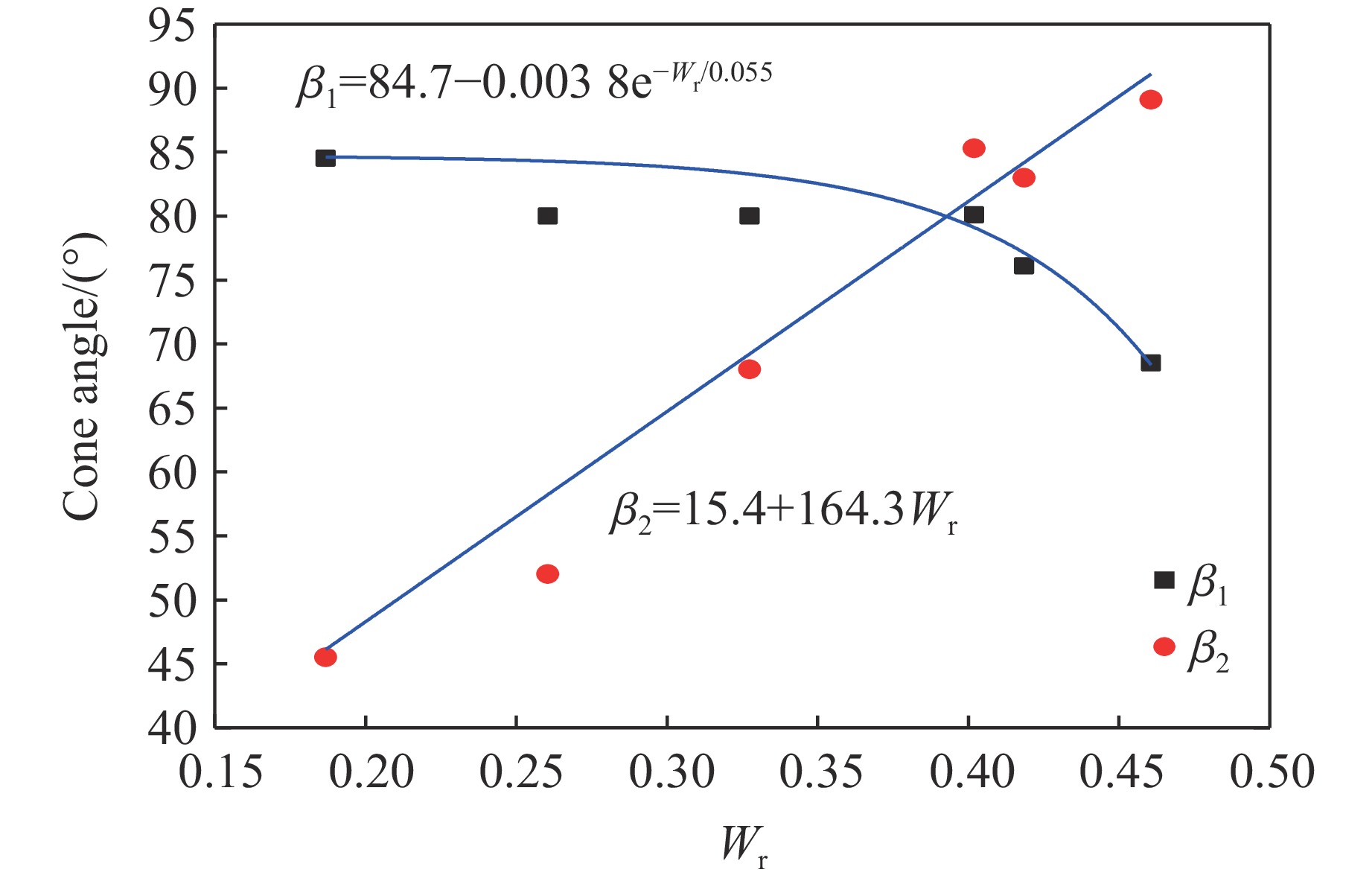
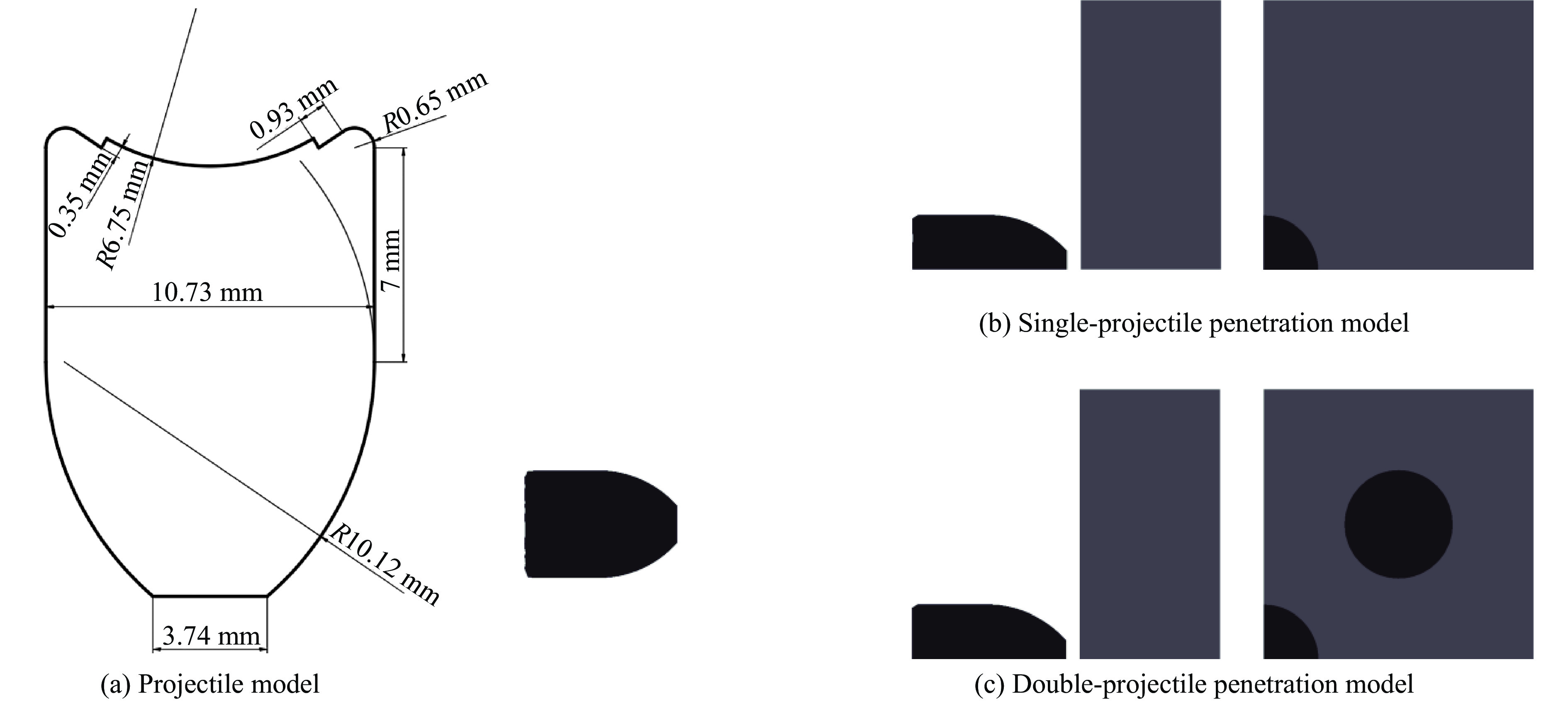
To investigate the influence of backplate mechanical properties on the formation mechanism of ceramic cones in boron carbide ceramic composite armor, four typical backplate materials, including 6061 aluminum alloy, 7075 aluminum alloy, T300 carbon fiber board, and ultra-high molecular weight polyethylene (UHMWPE) were selected. A combination of ballistic impact experiments conducted via a one-stage light gas gun and numerical simulations performed with LS-DYNA was adopted to systematically study the effects of backplate yield strength, stiffness, and wave impedance on the morphology and evolution of ceramic cones. The results indicate that: the load transfer from the ceramic cone to the backplate is not solely dependent on a single outer cone but is achieved through the synergistic action of multiple cracks, including the outer and inner cones; the yield strength of the backplate has no significant effect on the crack propagation of the main cone; regarding stiffness, the outer cone angle decreases linearly with increasing elastic modulus, while the inner cone angle increases exponentially; wave impedance alters the internal stress field of the ceramic by modulating stress wave reflection/transmission, resulting in a linear increase in the inner cone angle and an exponential decrease in the outer cone angle with increasing impedance.


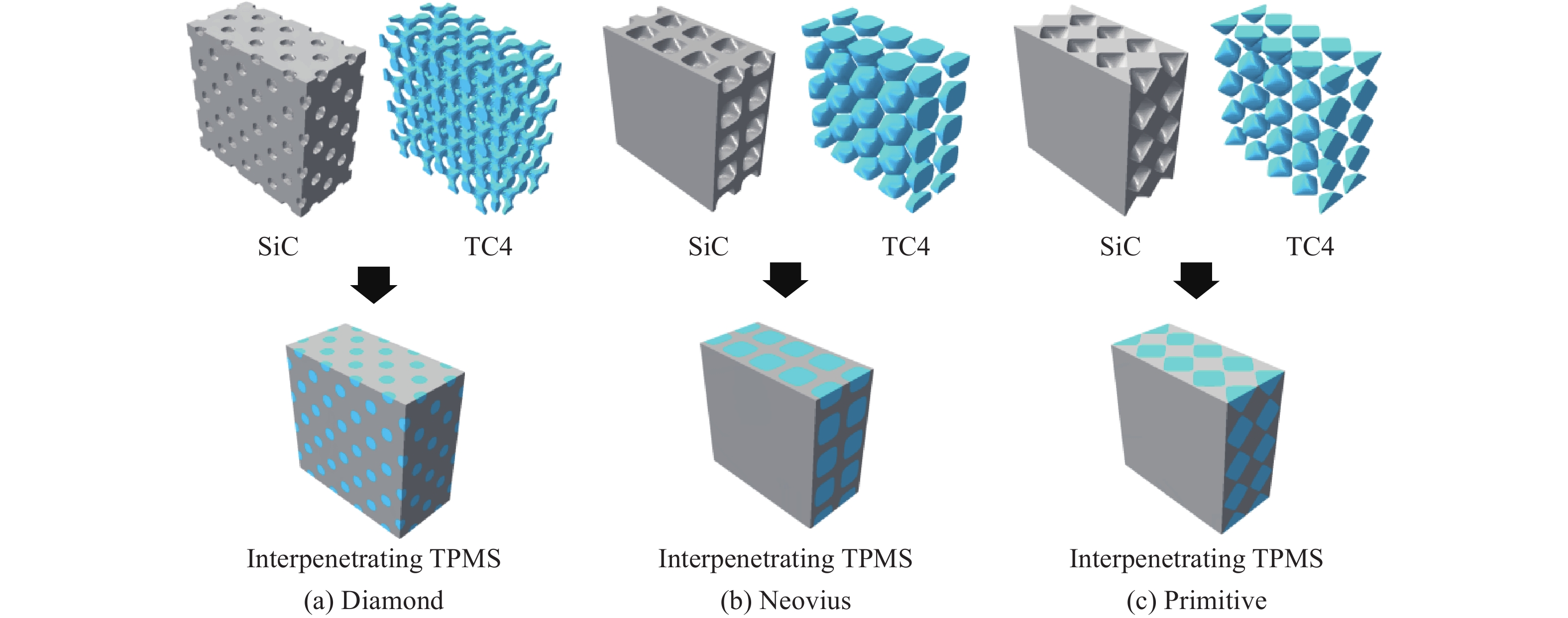
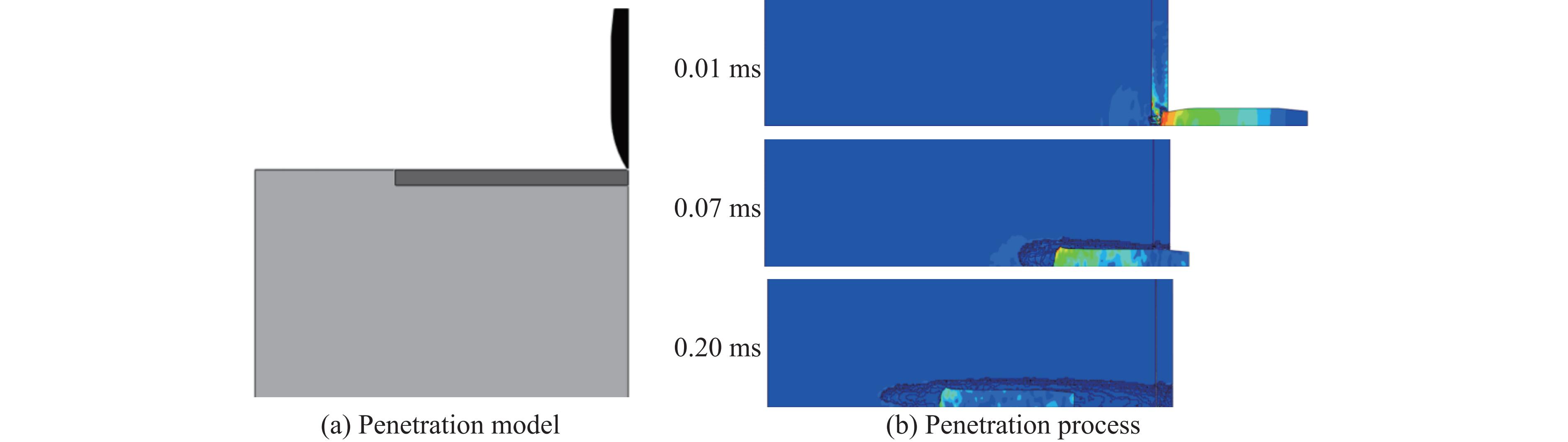
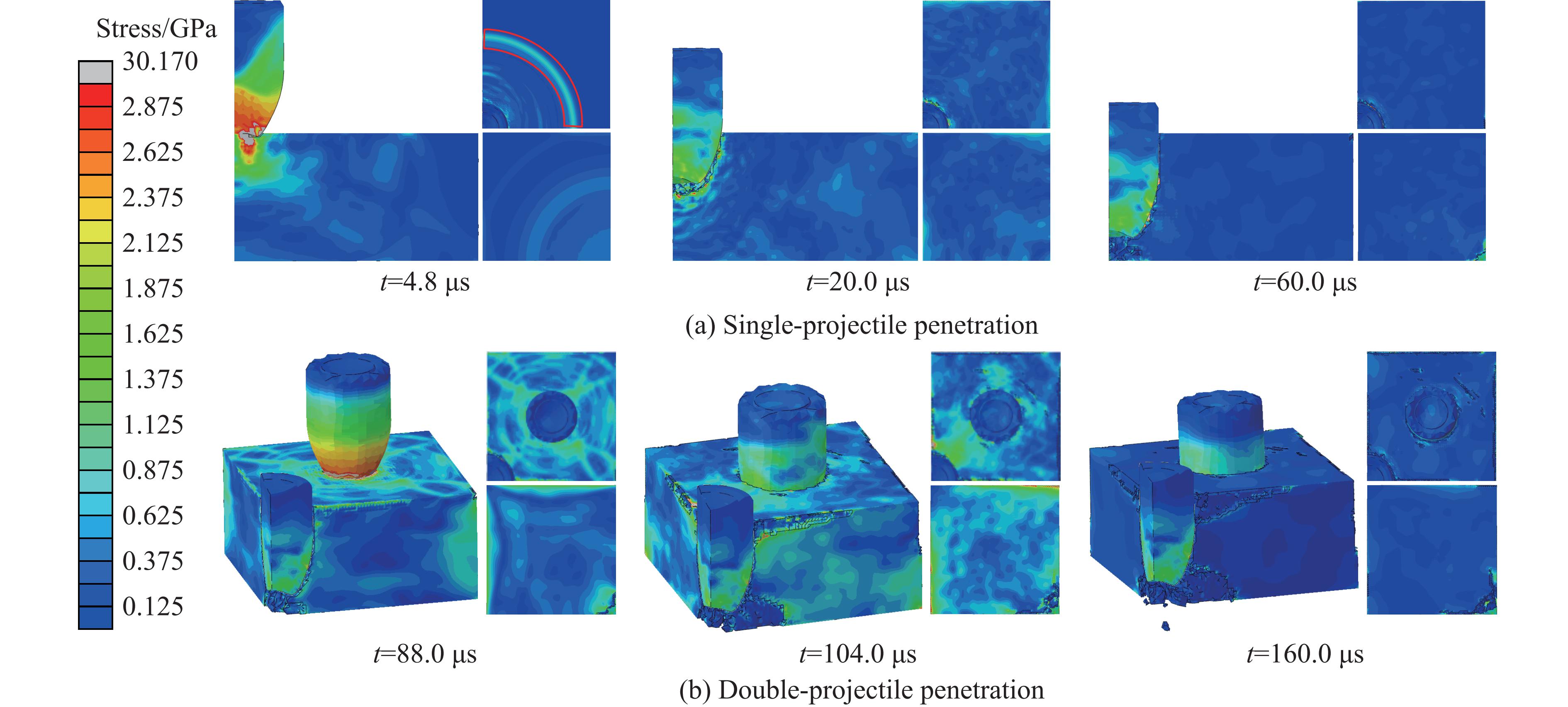
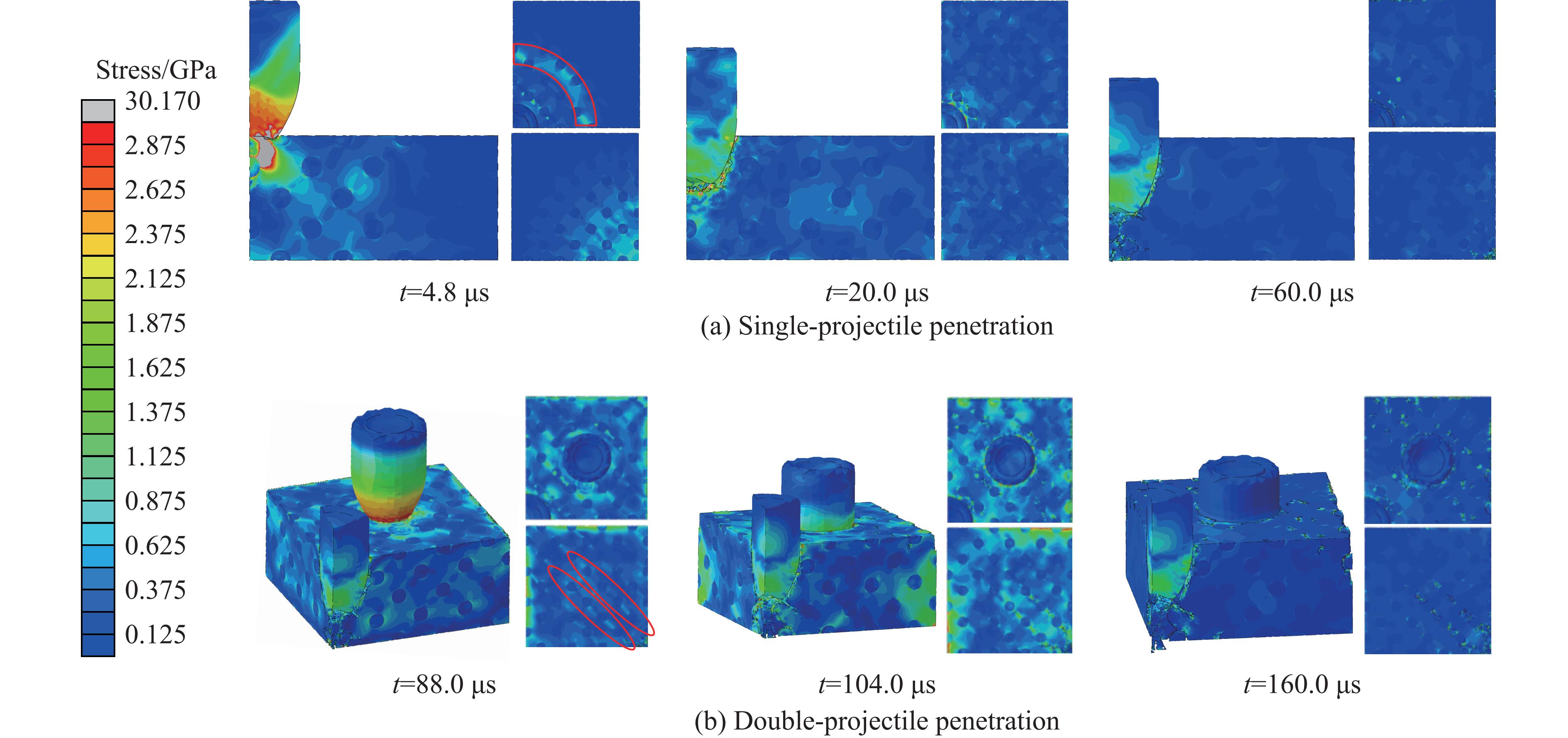
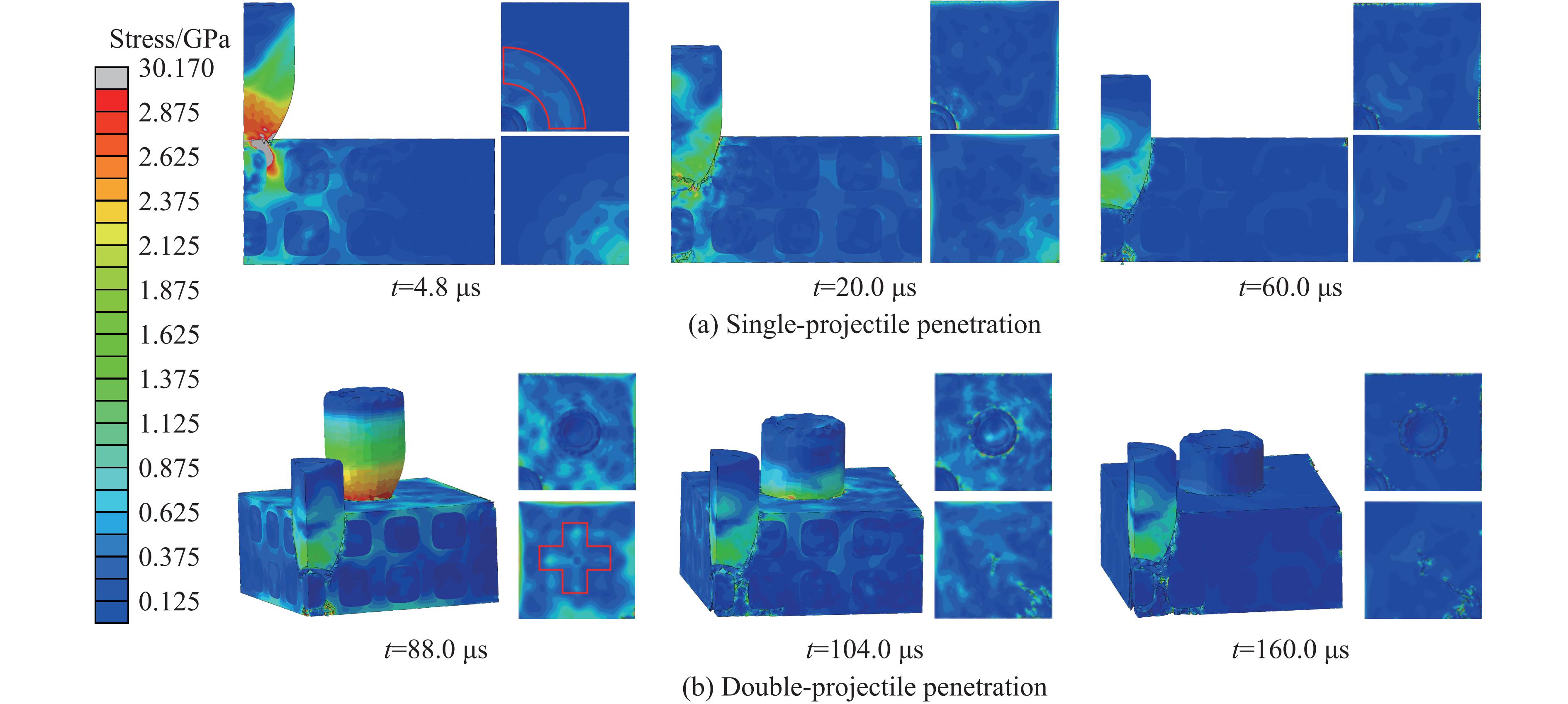
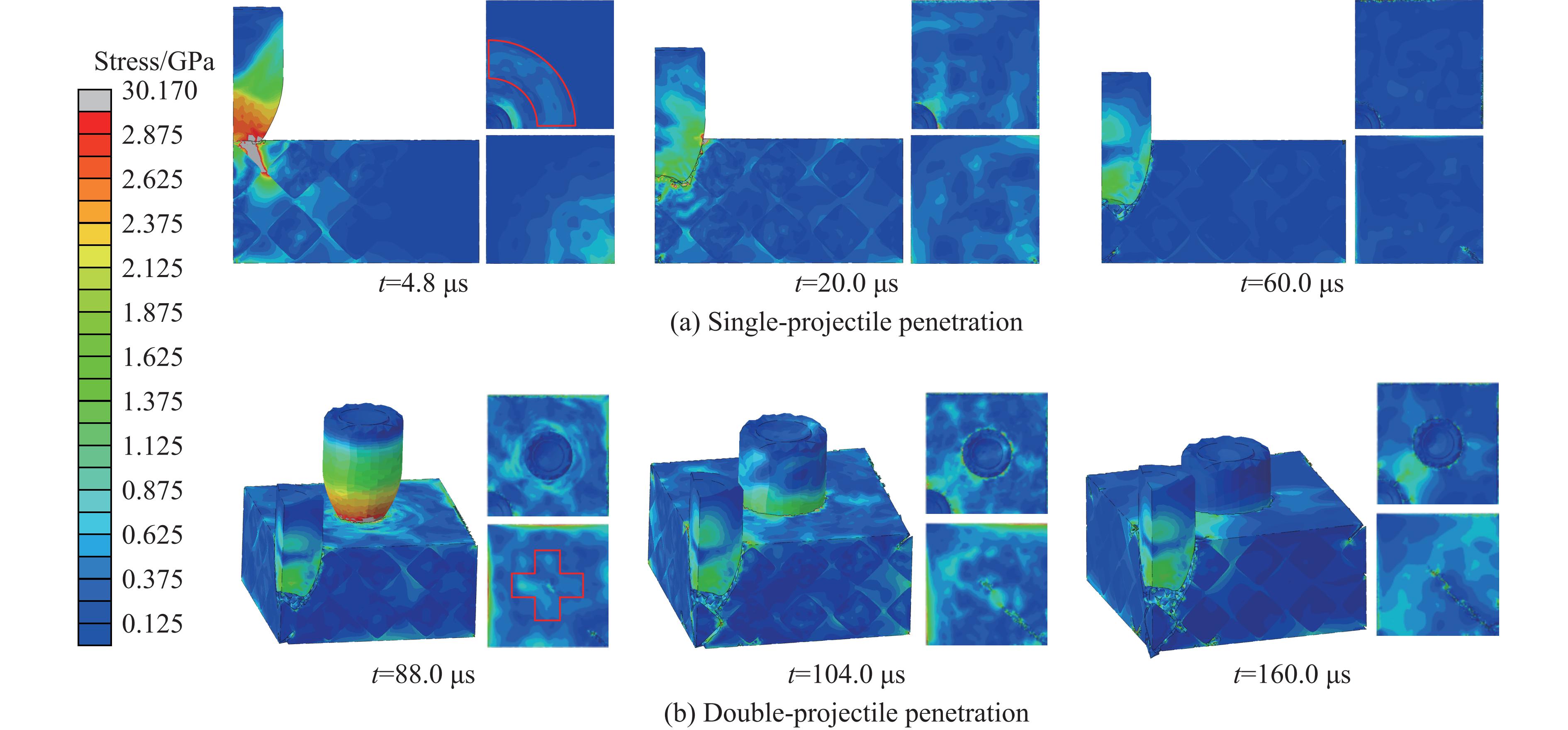
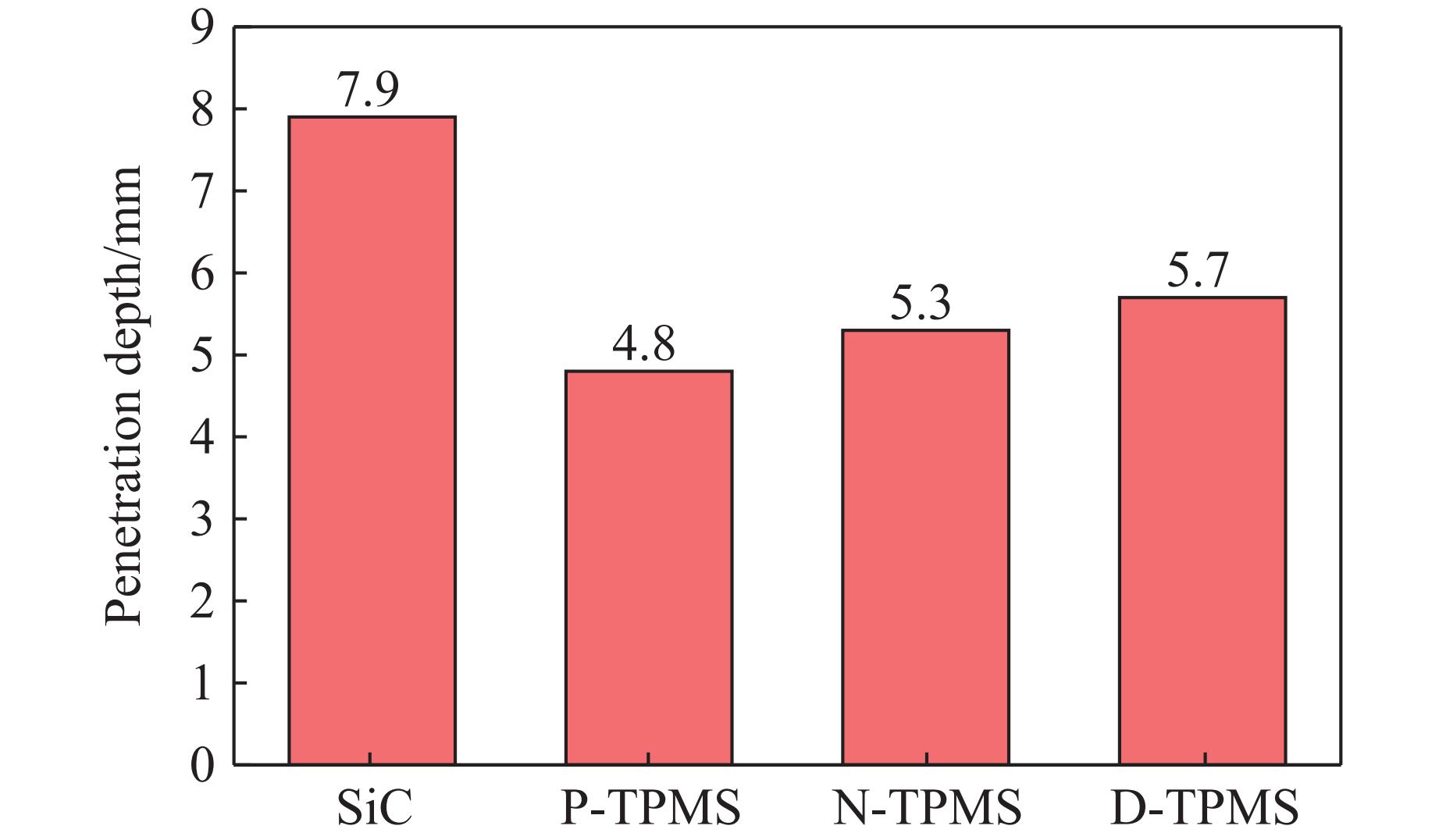
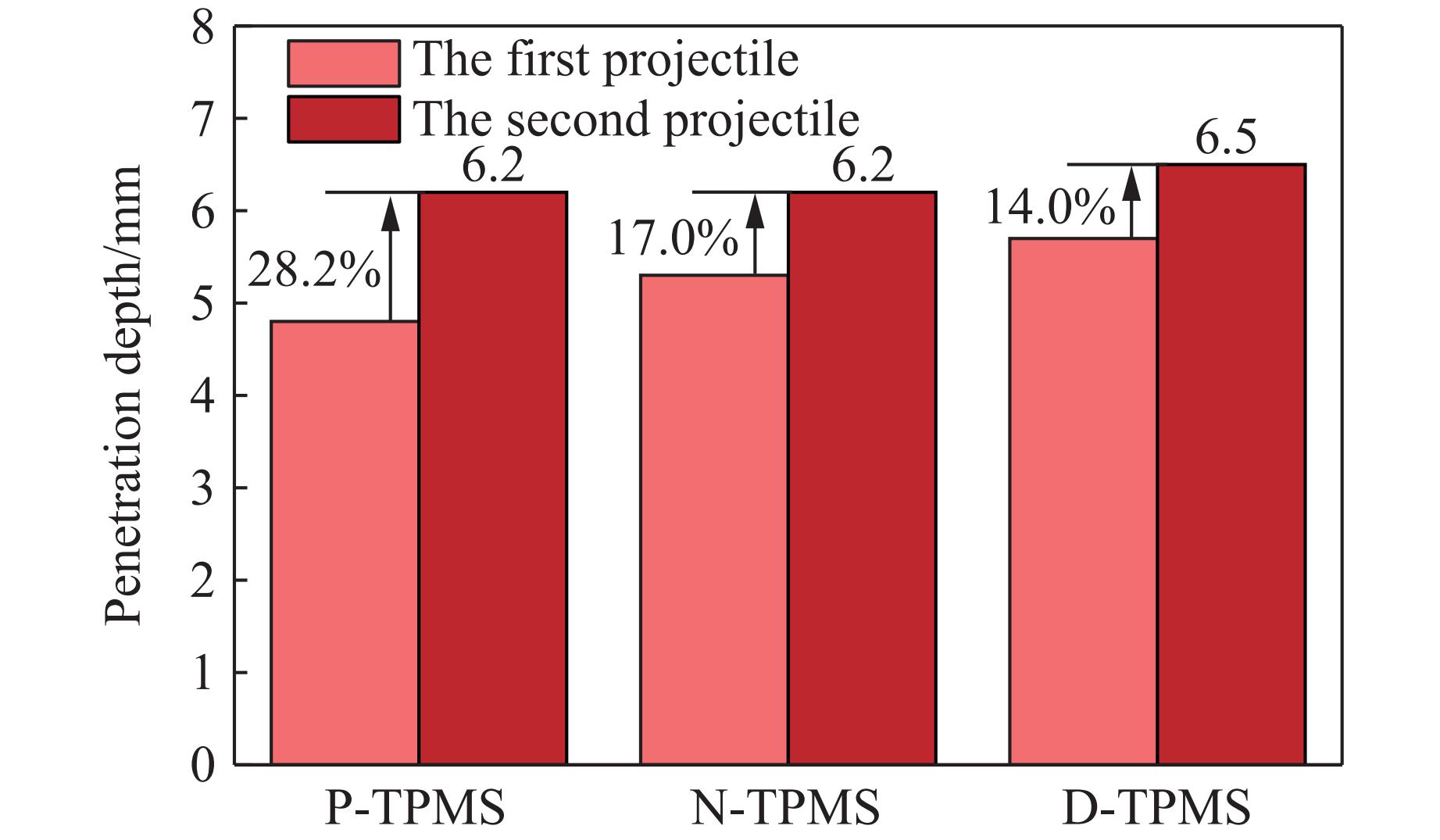
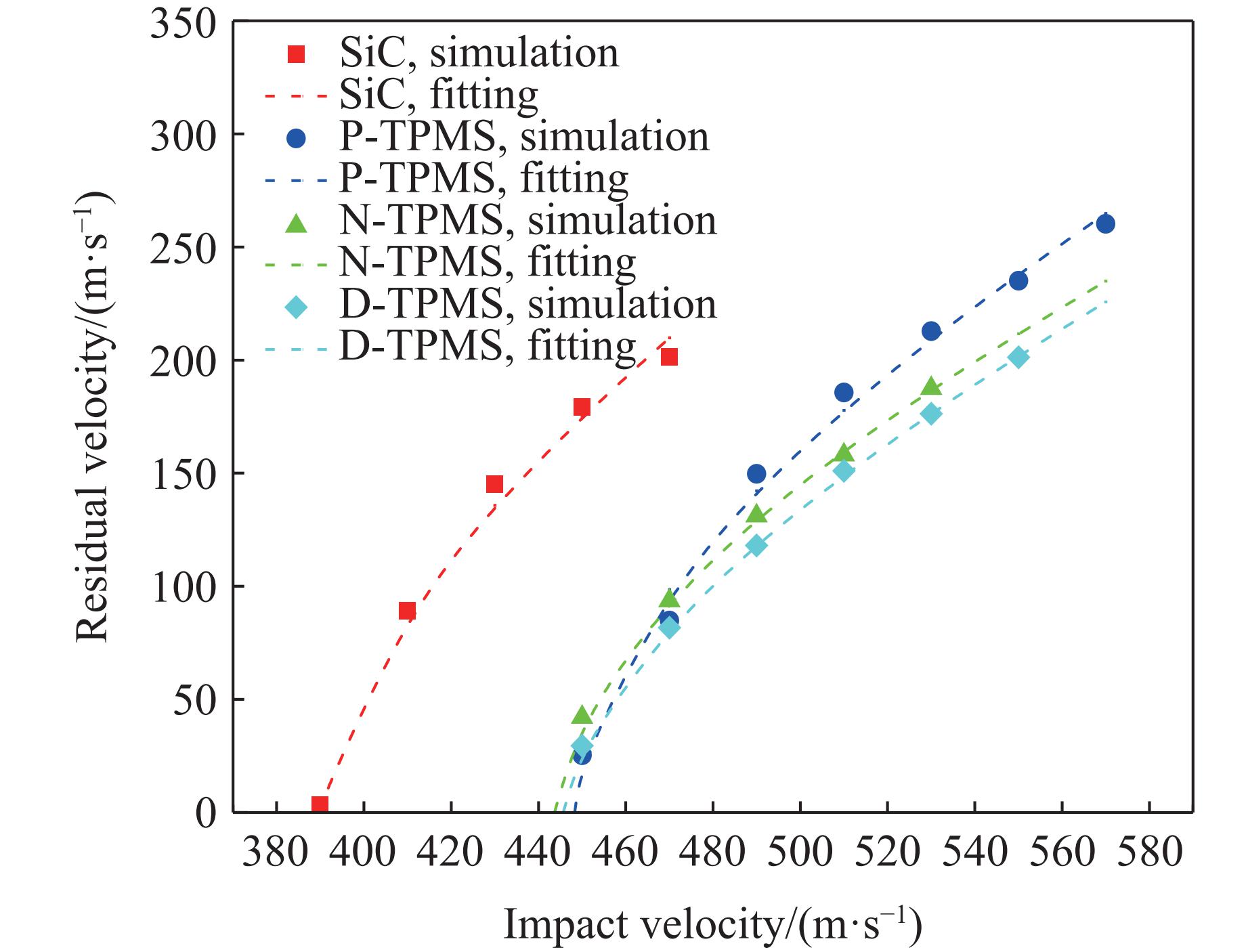
Ceramic/metal composite materials were widely used in national defense, military industry, and aerospace fields as lightweight impact-resistant structures with high specific strength and high energy absorption efficiency. With the development of 3D printing technology, it has become possible to fabricate complex lattice structures based on triply periodic minimal surfaces (TPMS). In this paper, an interpenetrating TPMS ballistic composite structure composed of silicon carbide (SiC) ceramic and titanium alloy (TC4) is designed. A series of numerical simulations are carried out under single-projectile and double-projectile penetration conditions using ABAQUS software. The damage modes, penetration depth, and ballistic limit velocity of the proposed structure and pure SiC target plate are compared and analyzed. The simulation results show that different interpenetrating TPMS structures exhibit distinct damage and failure modes. The three-dimensional topological configuration restrains crack propagation inside the ceramic, resulting in slighter overall damage than the pure SiC target plate. The damage caused by the second projectile further propagates along the penetration region of the first projectile, and accompanied by an increase in penetration depth. Compared with the pure SiC target plate, the three interpenetrating TPMS targets present smaller penetration depth and higher ballistic limit velocity. When the projectile can perforate the target plate, the primitive (P-type) structure shows better ballistic performance against low-velocity projectiles, while the diamond (D-type) structure is superior against high-velocity projectiles. It is demonstrated that the interpenetrating TPMS targets possess better ballistic performance than pure SiC at the same area density. The technical support and theoretical basis can be provided for the design of novel lightweight ceramic armor in this study.

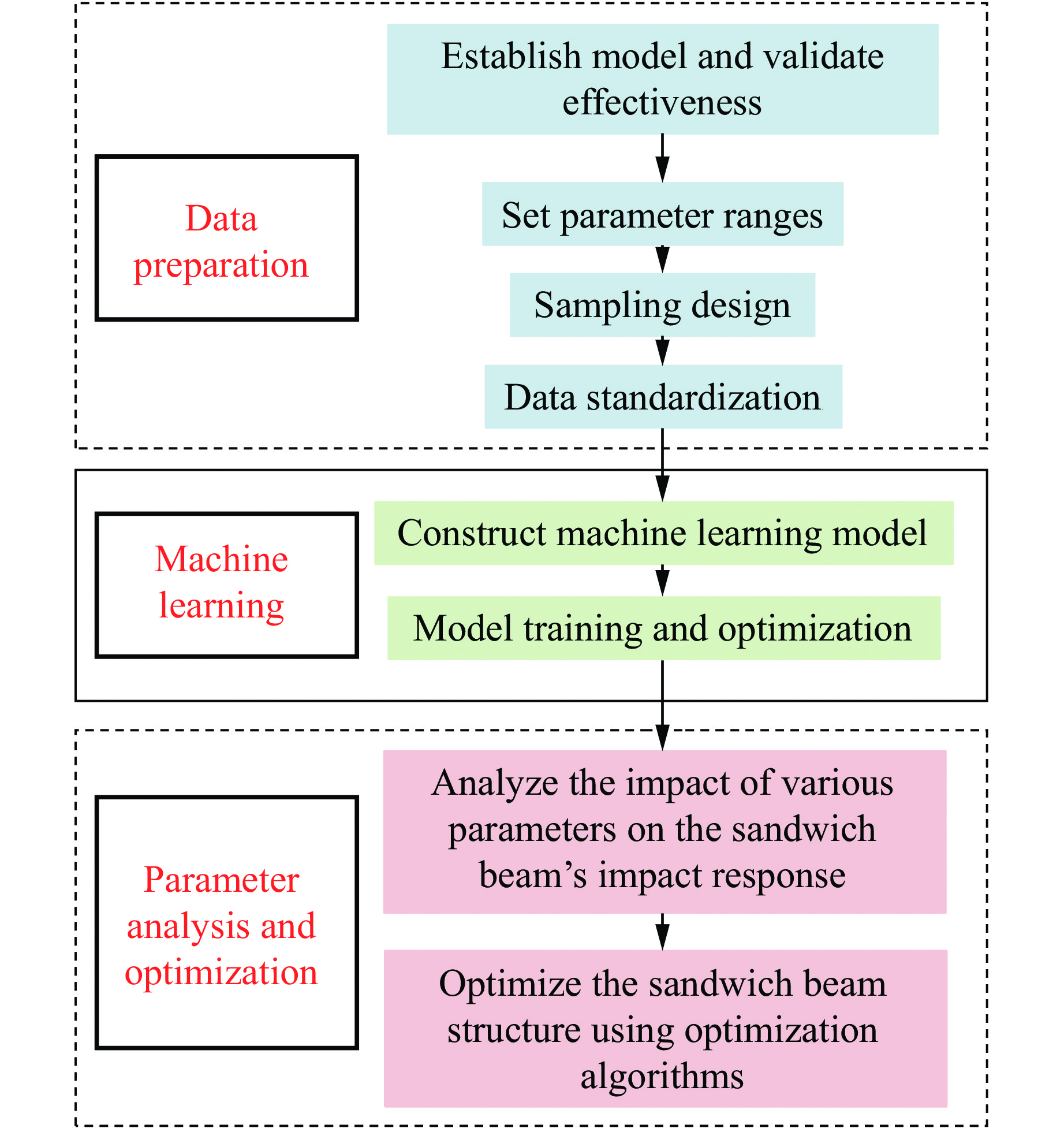
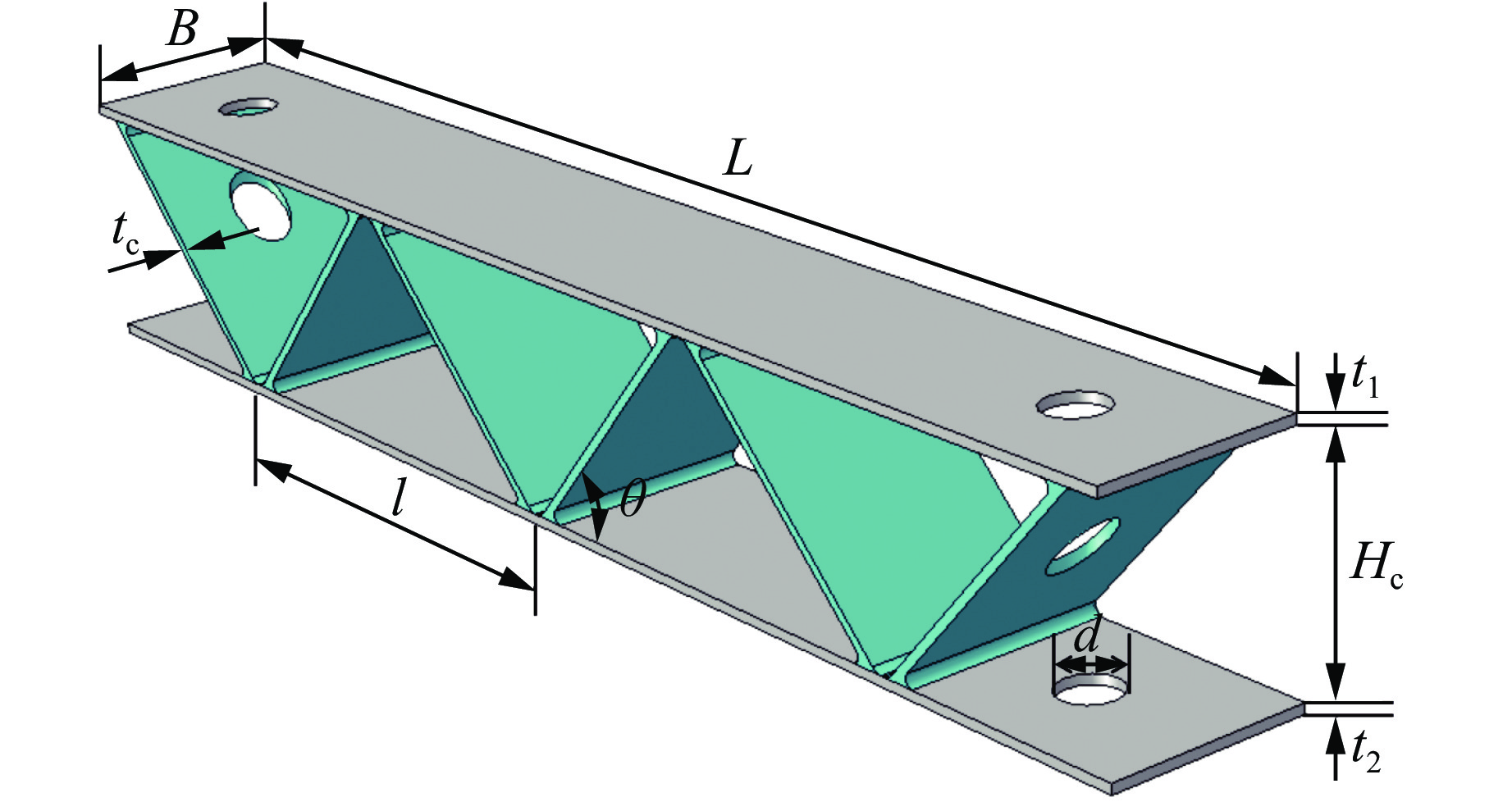
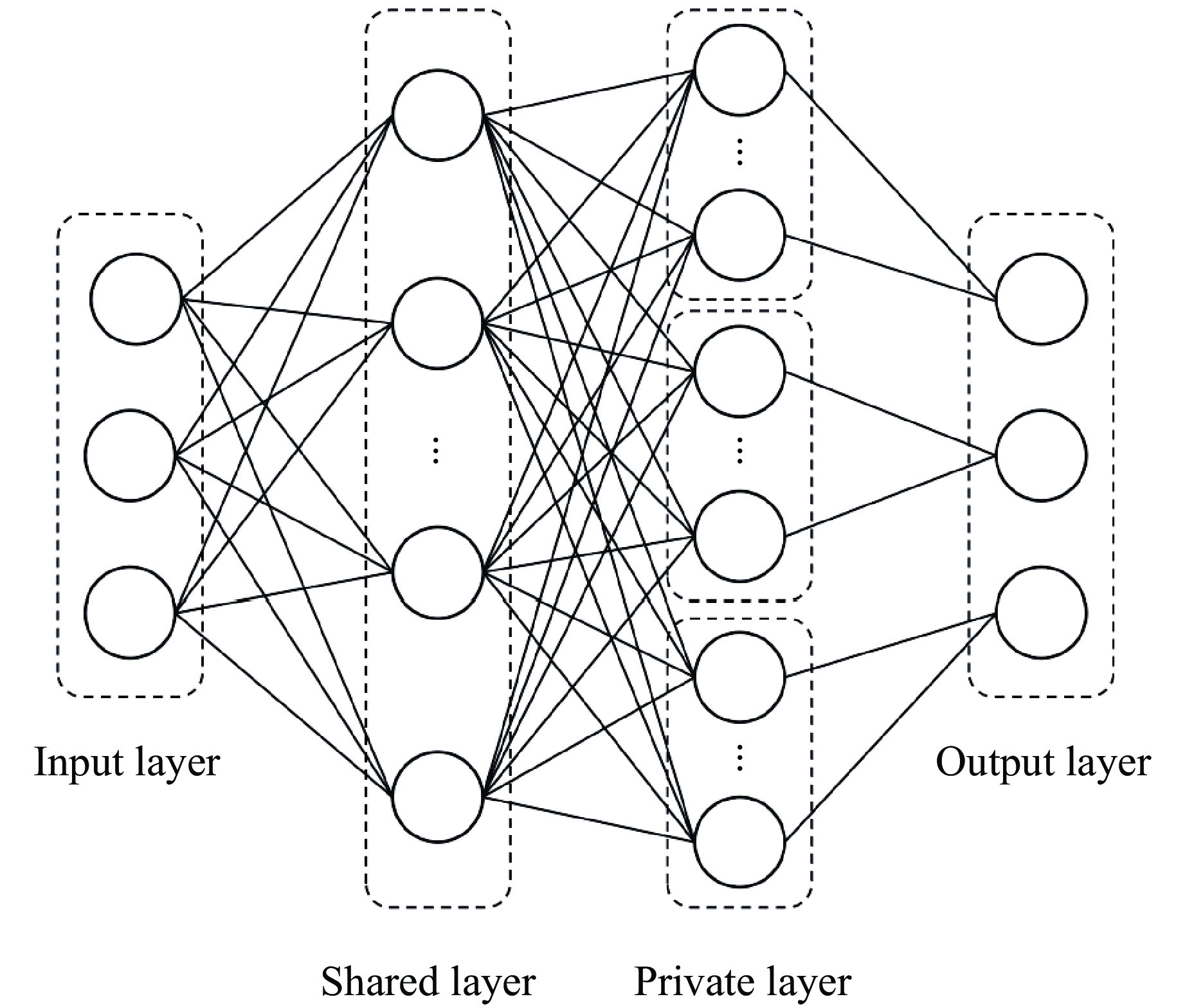
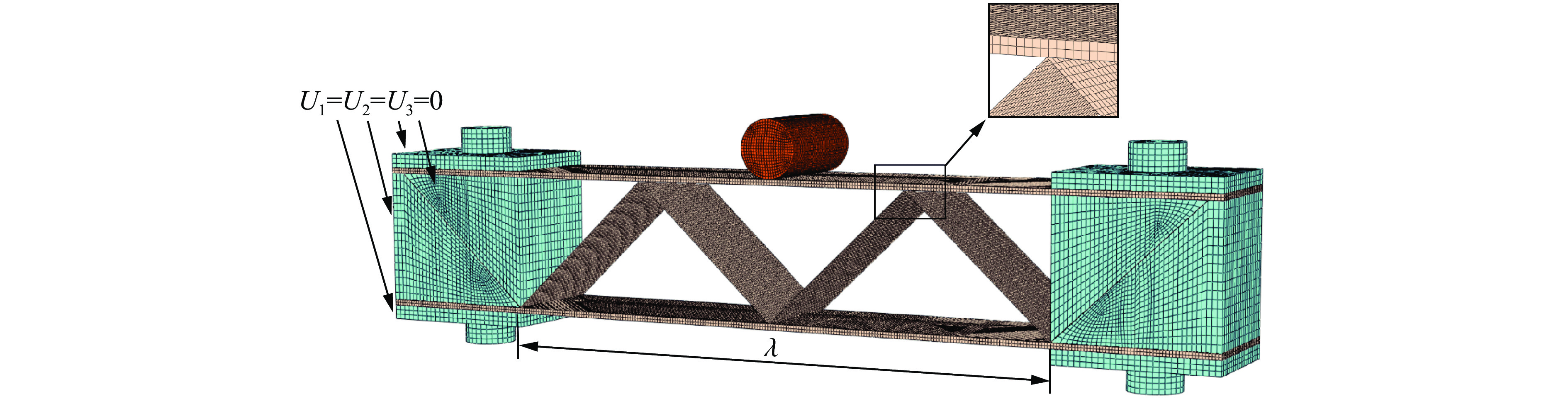
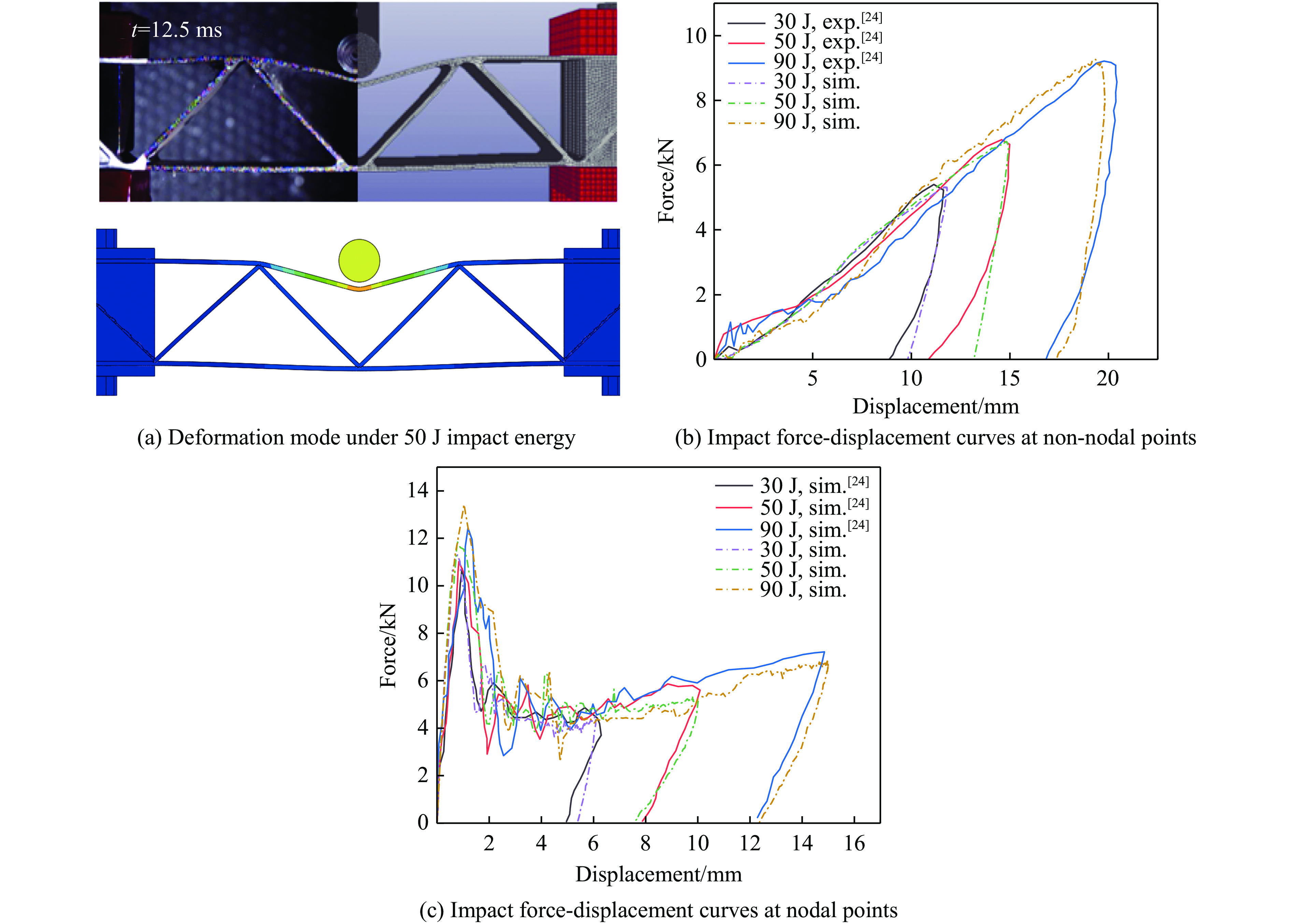
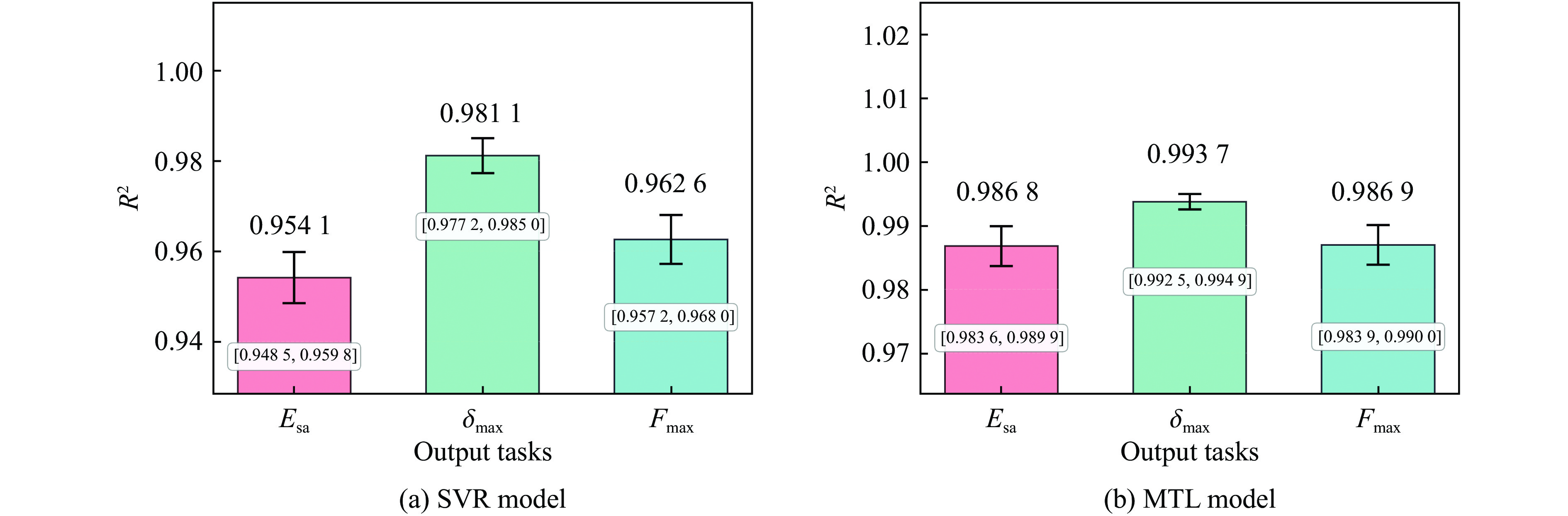
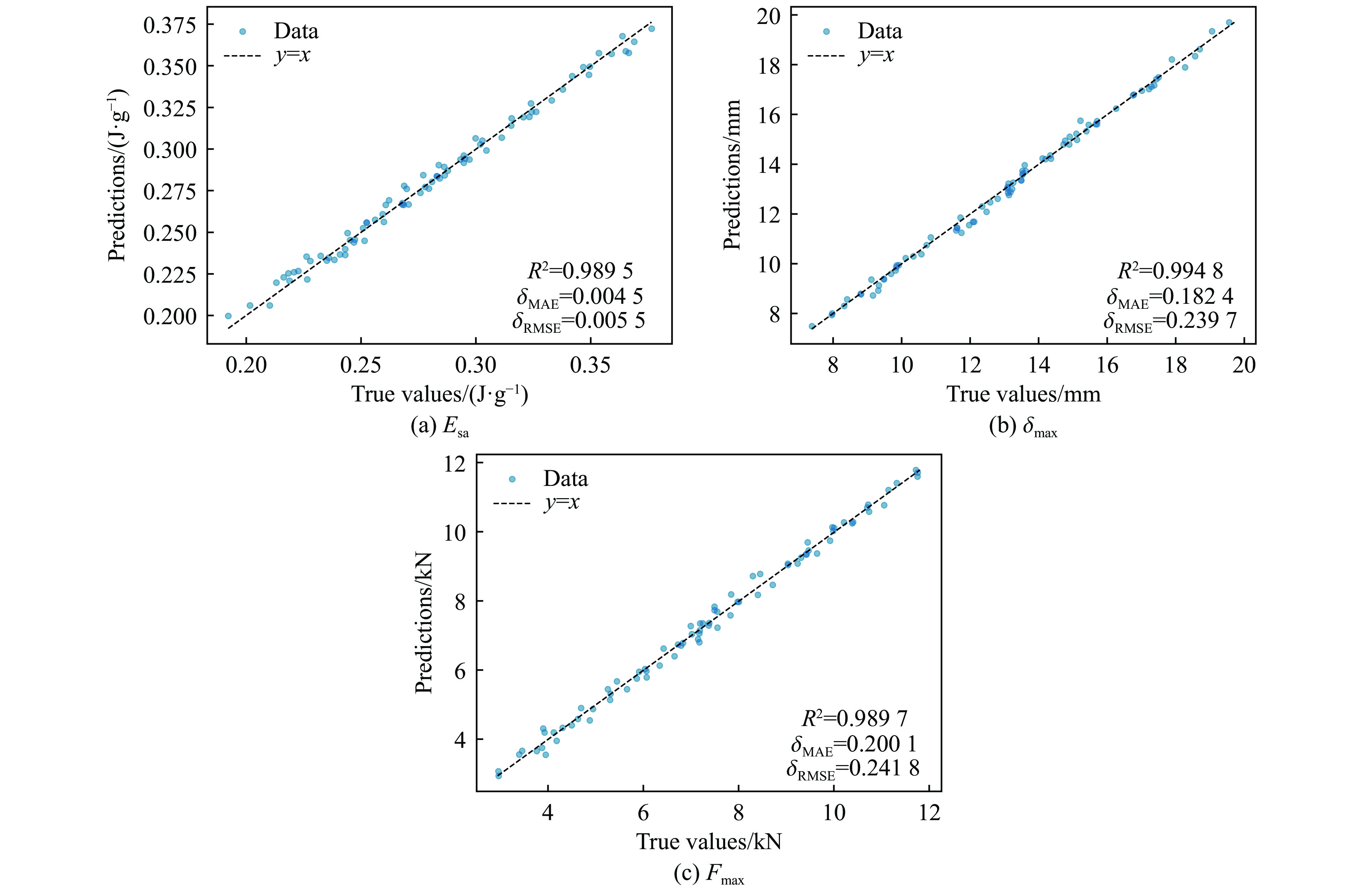
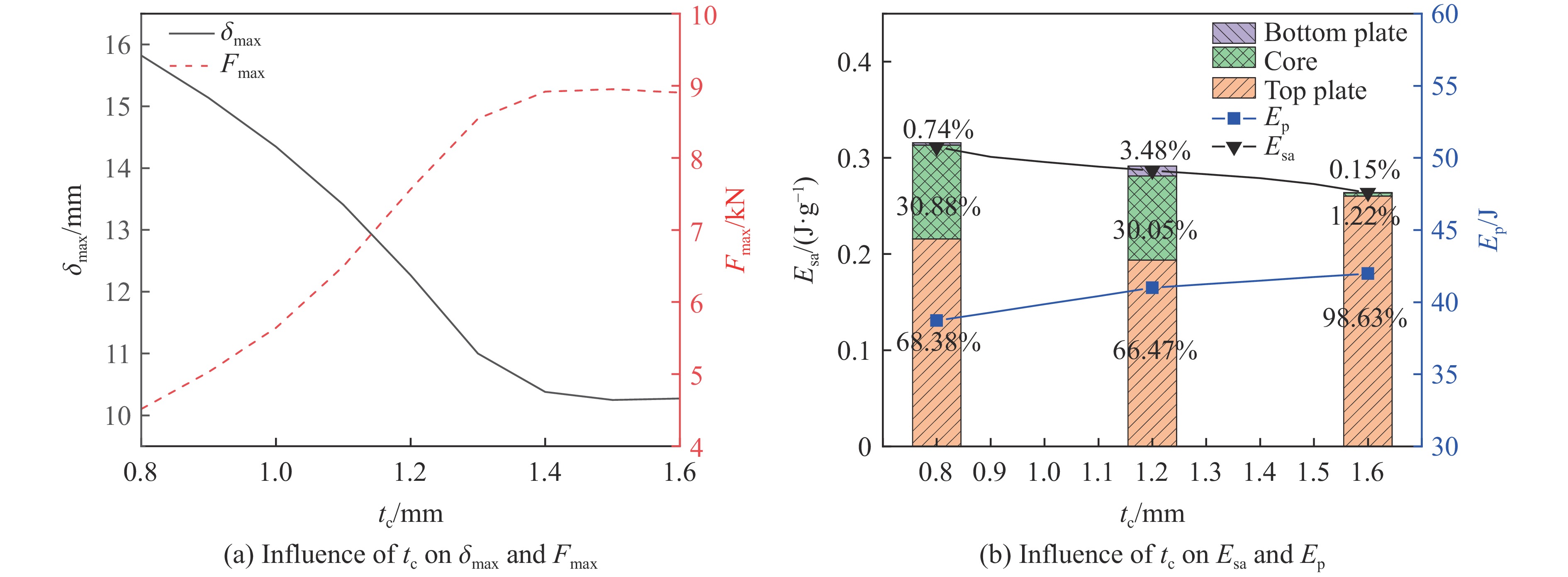
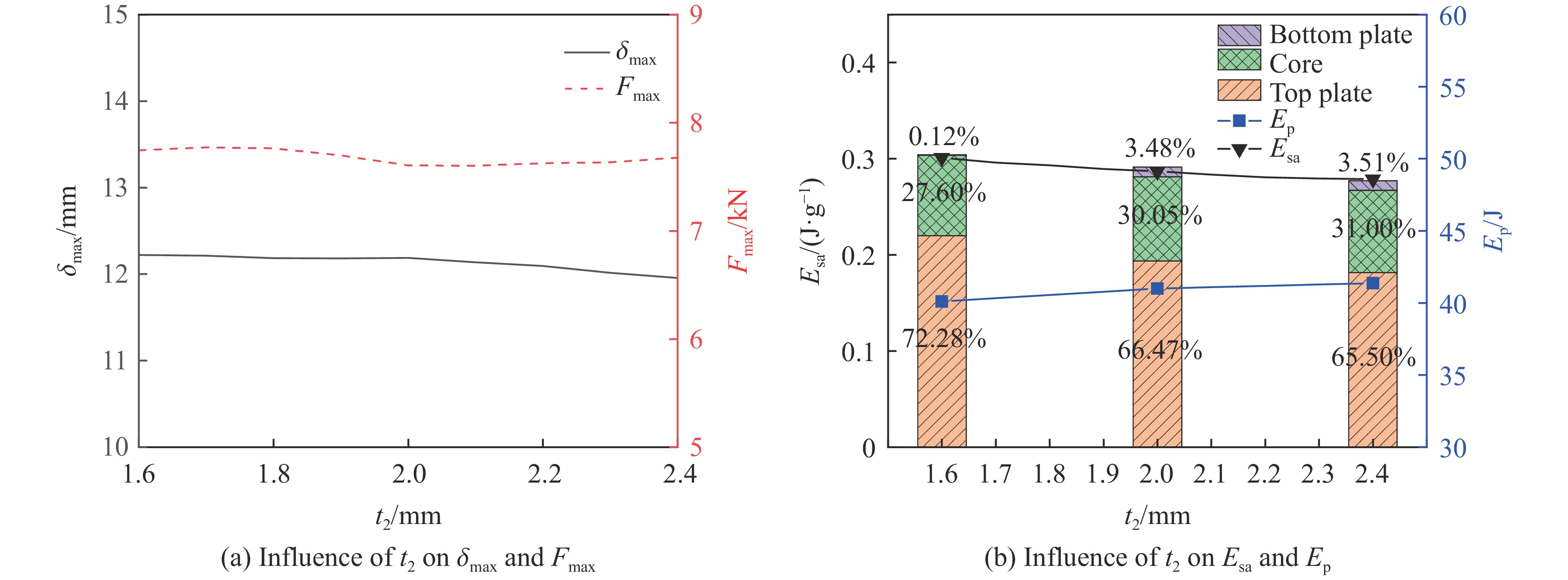
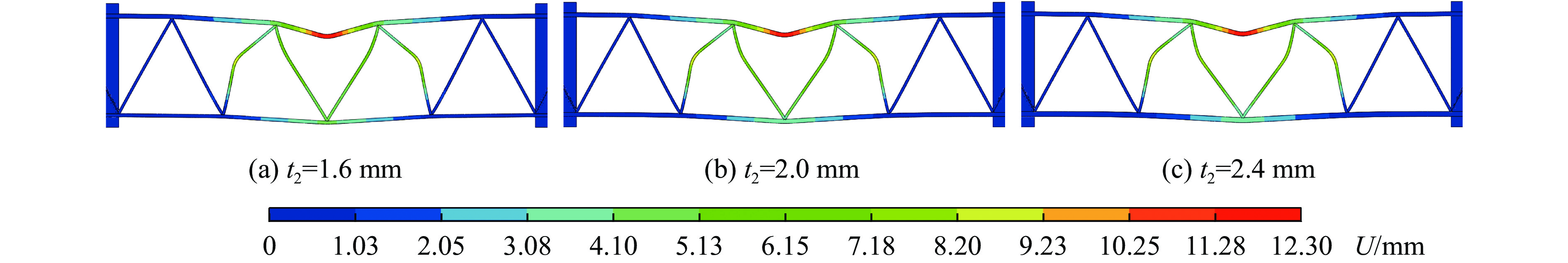
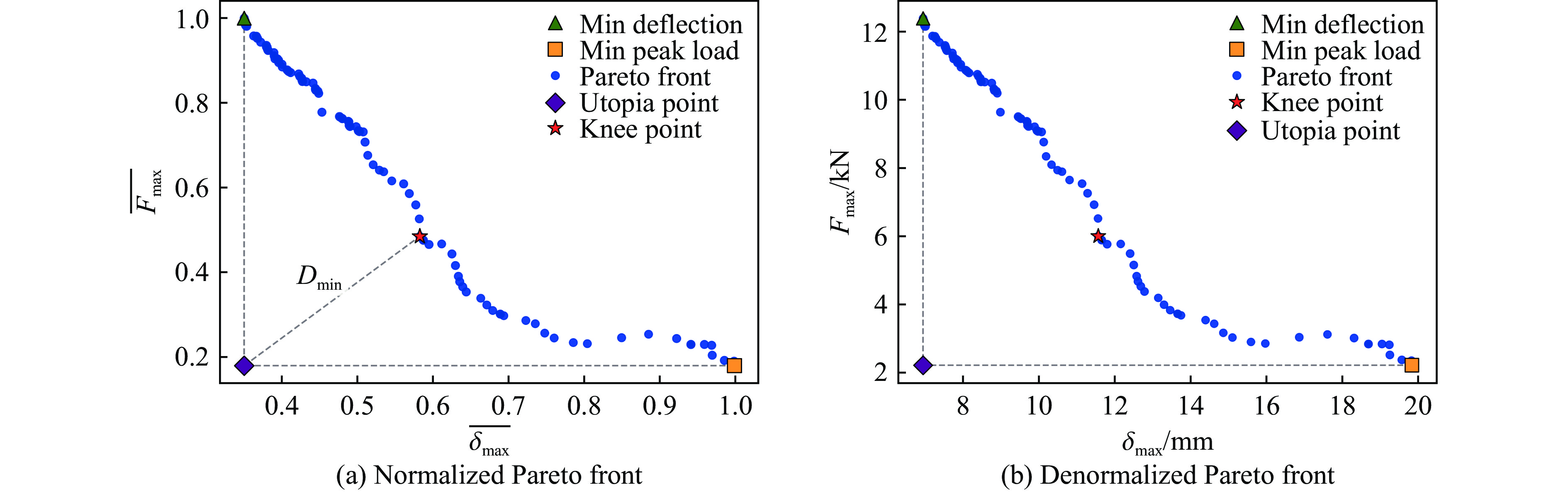
To enhance the prediction accuracy of low-velocity impact performance and improve the structural design efficiency of triangular corrugated sandwich beams, this paper proposes a machine learning modeling and optimization process for the impact response of sandwich beams based on a hard-parameter-sharing multi-task learning (MTL) framework. A sample dataset is generated using finite element models, and the rationality of the models is validated against existing experimental results. Subsequently, an MTL model is trained to simultaneously predict the structural specific energy absorption (SEA), the maximum deflection of the top panel, and the initial peak load. The results show that the MTL model optimized via Bayesian optimization demonstrates strong predictive performance under a 50 J impact energy condition. The predictions align well with the finite element simulation results, with the coefficient of determination
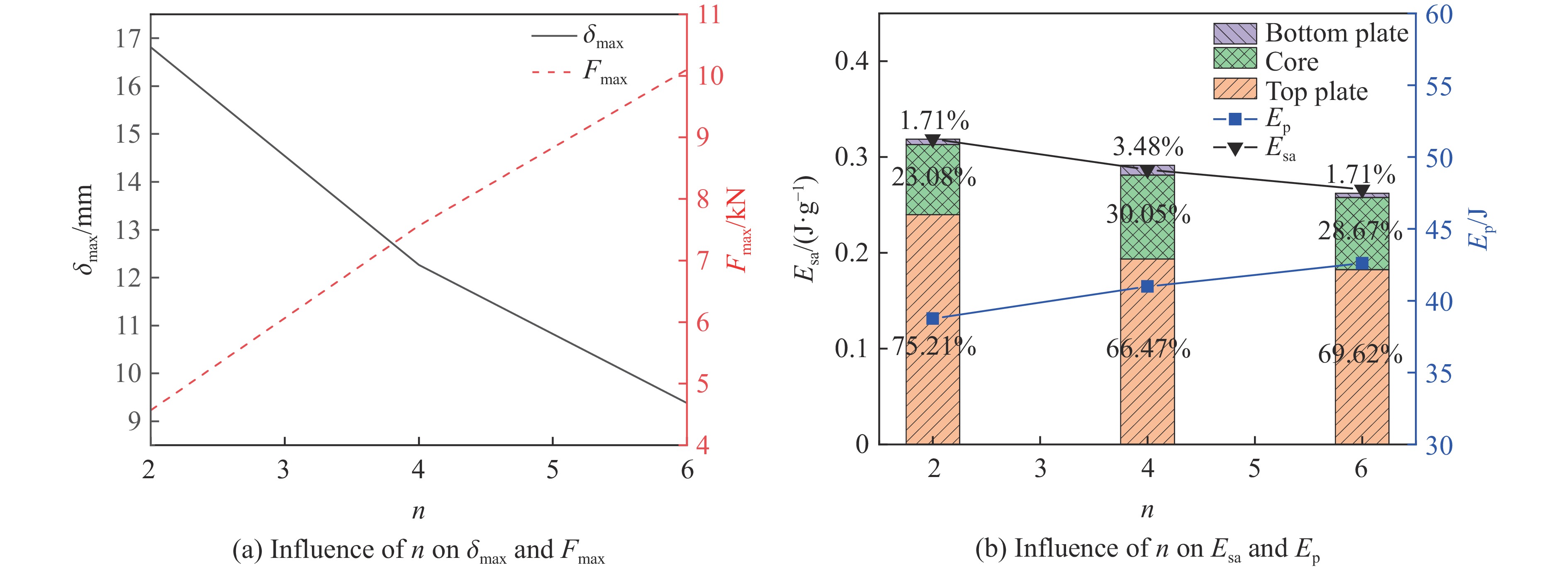
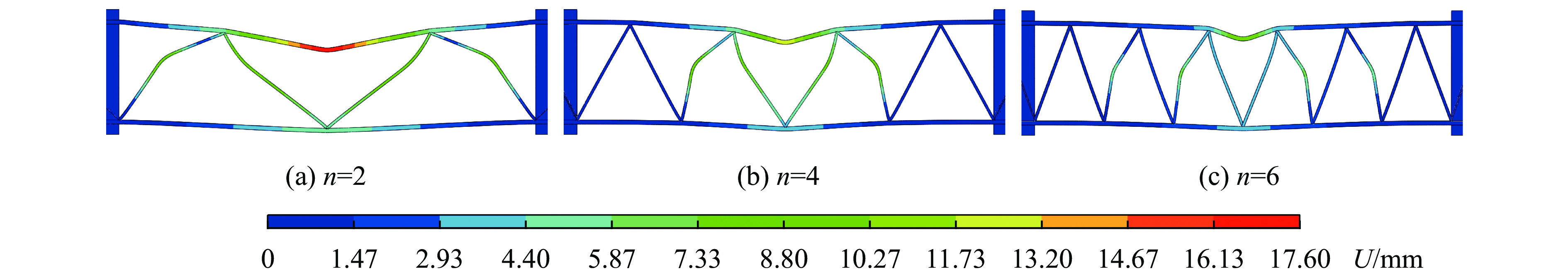
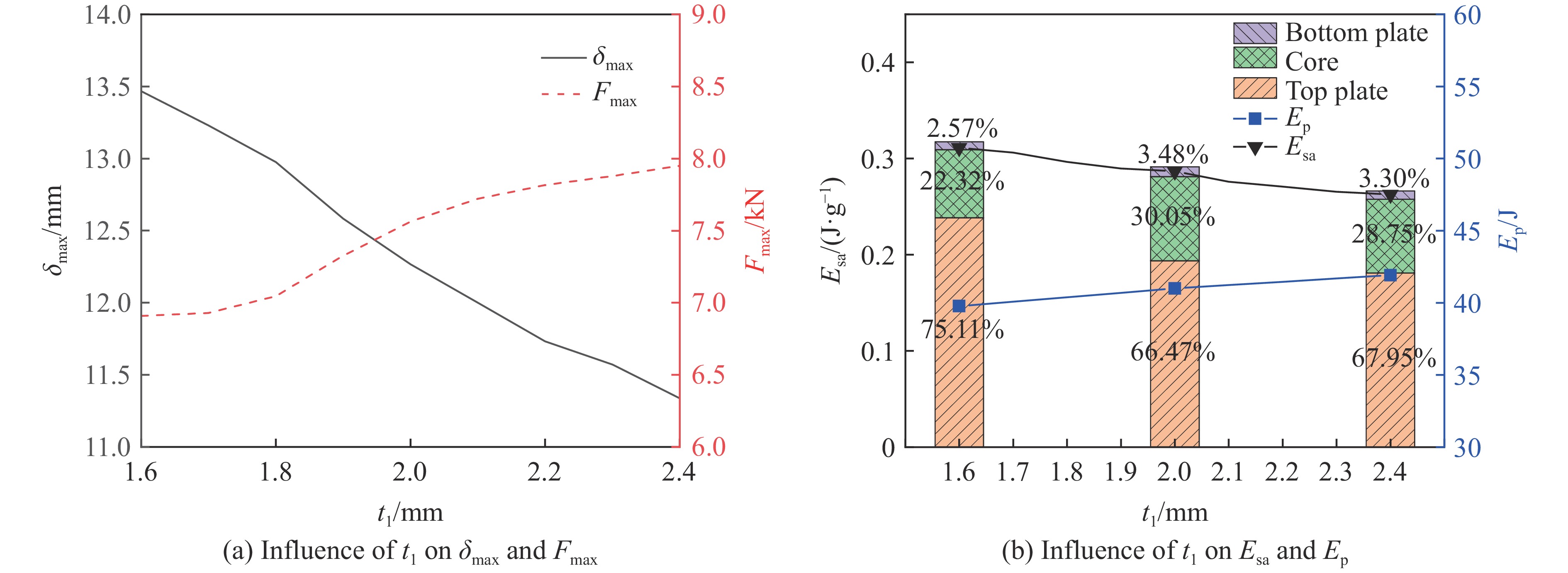
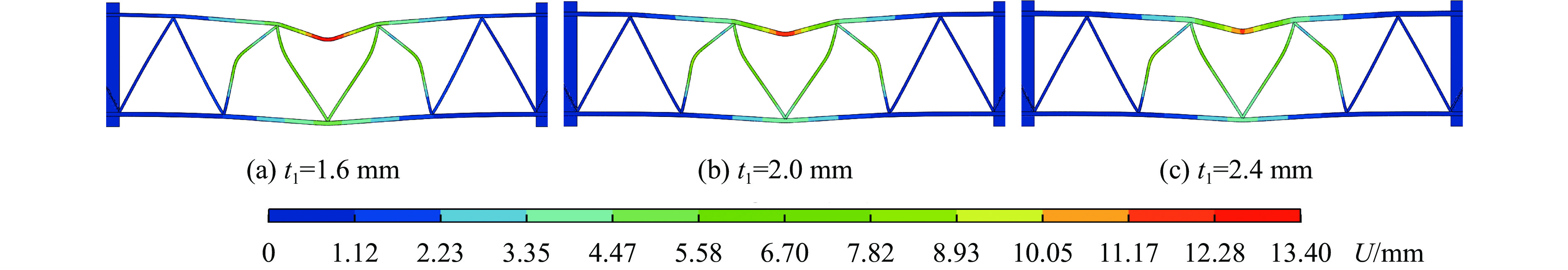

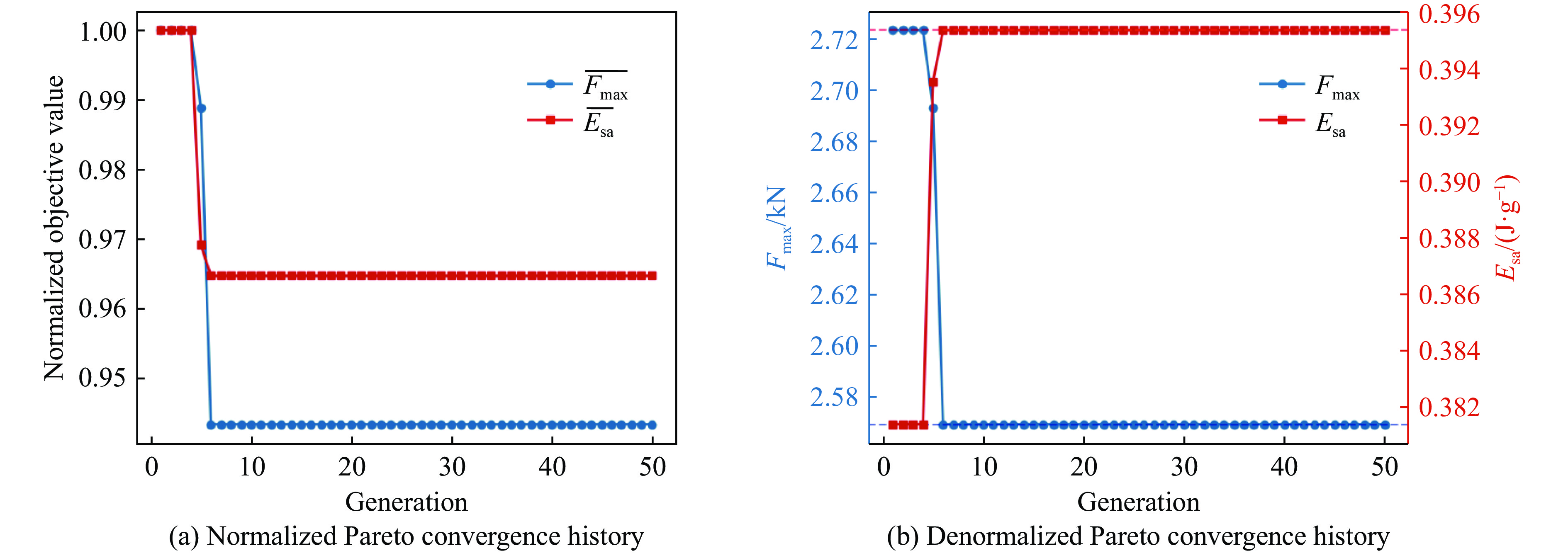
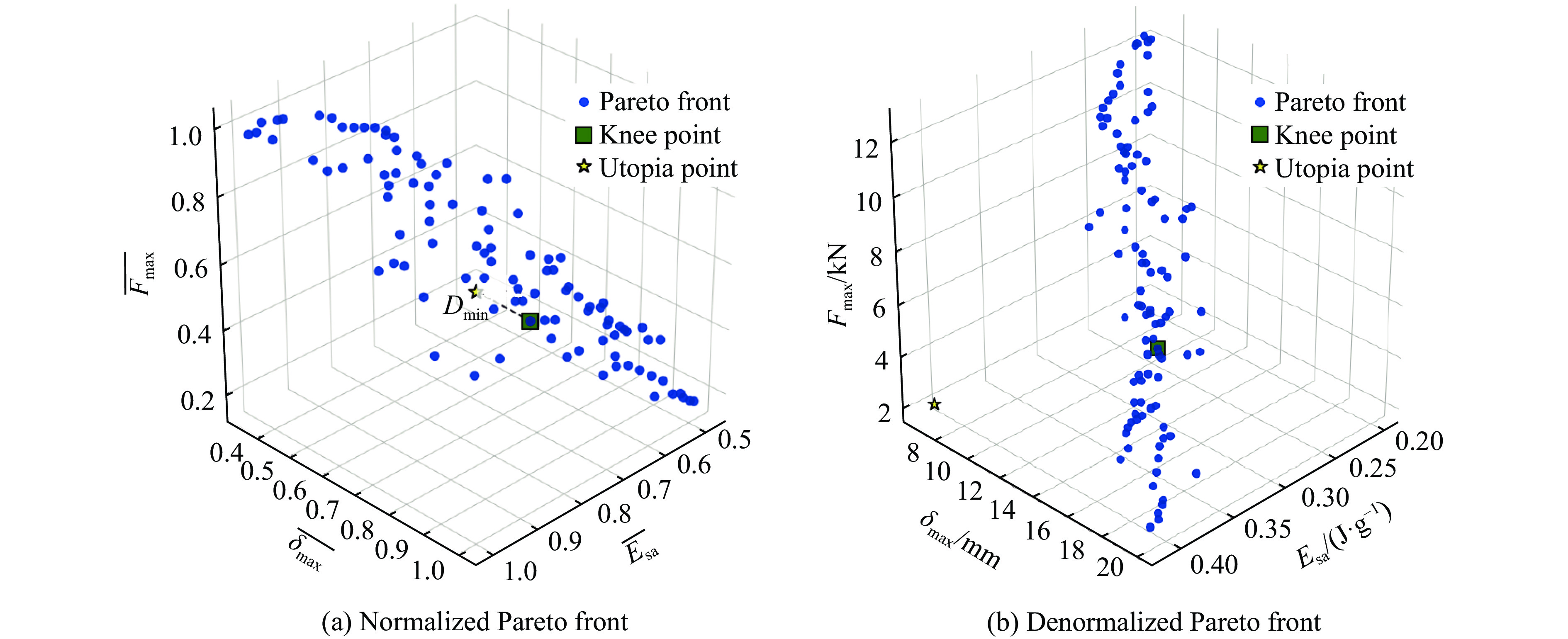
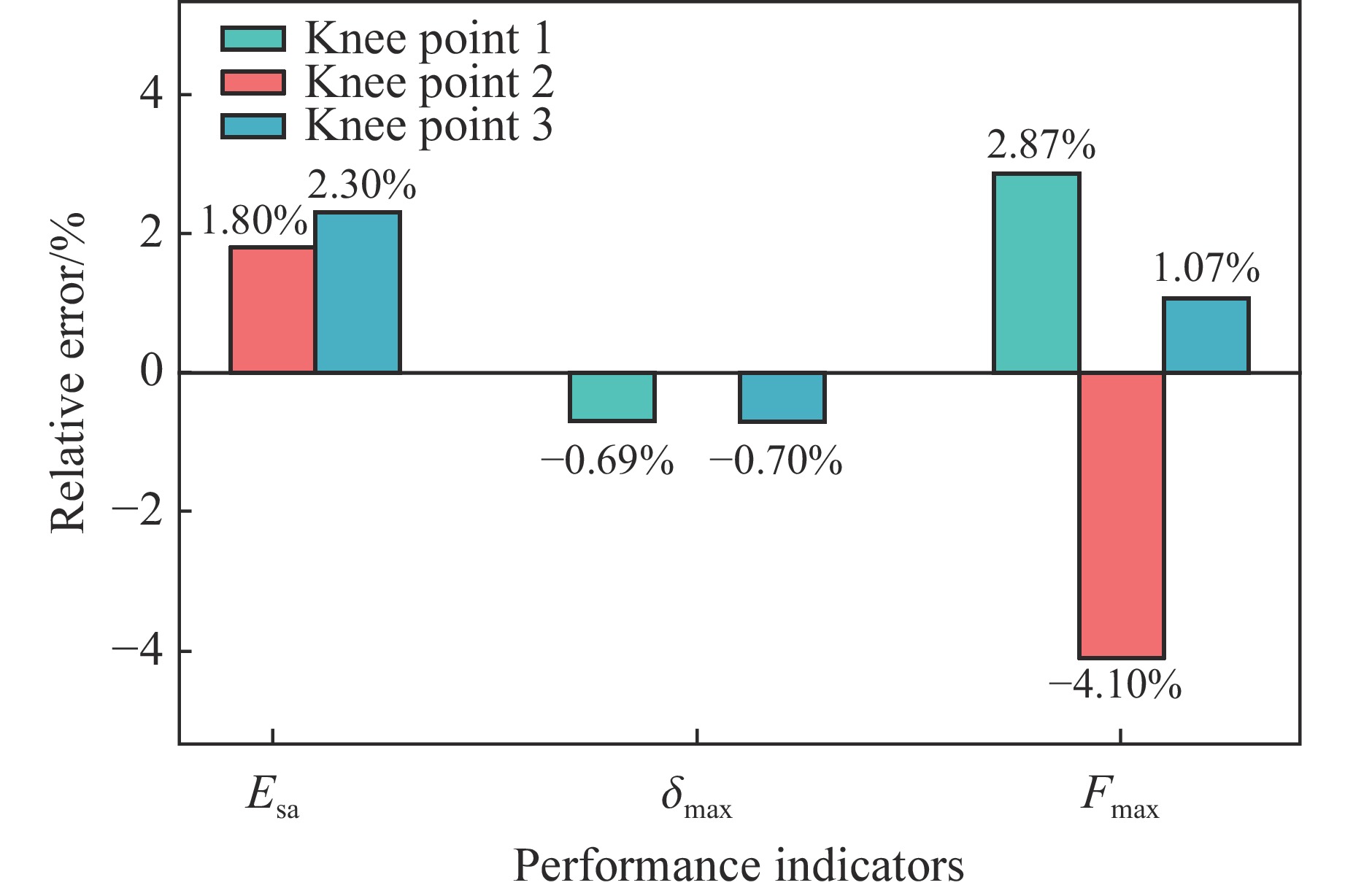
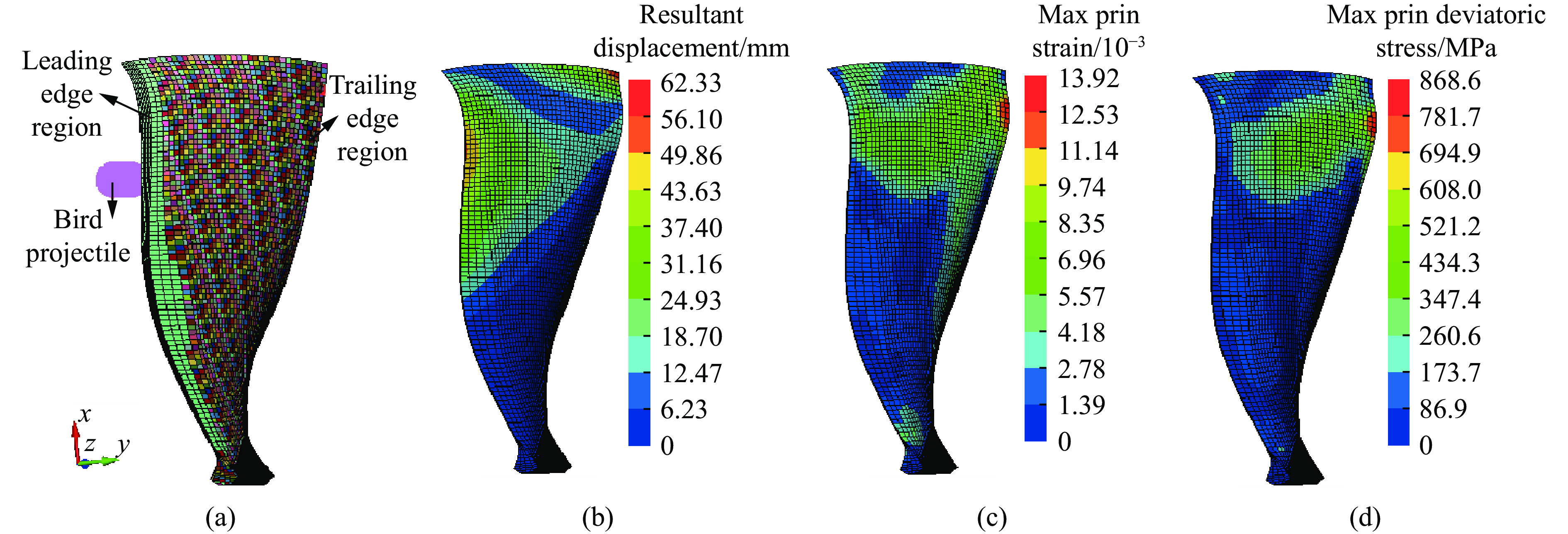
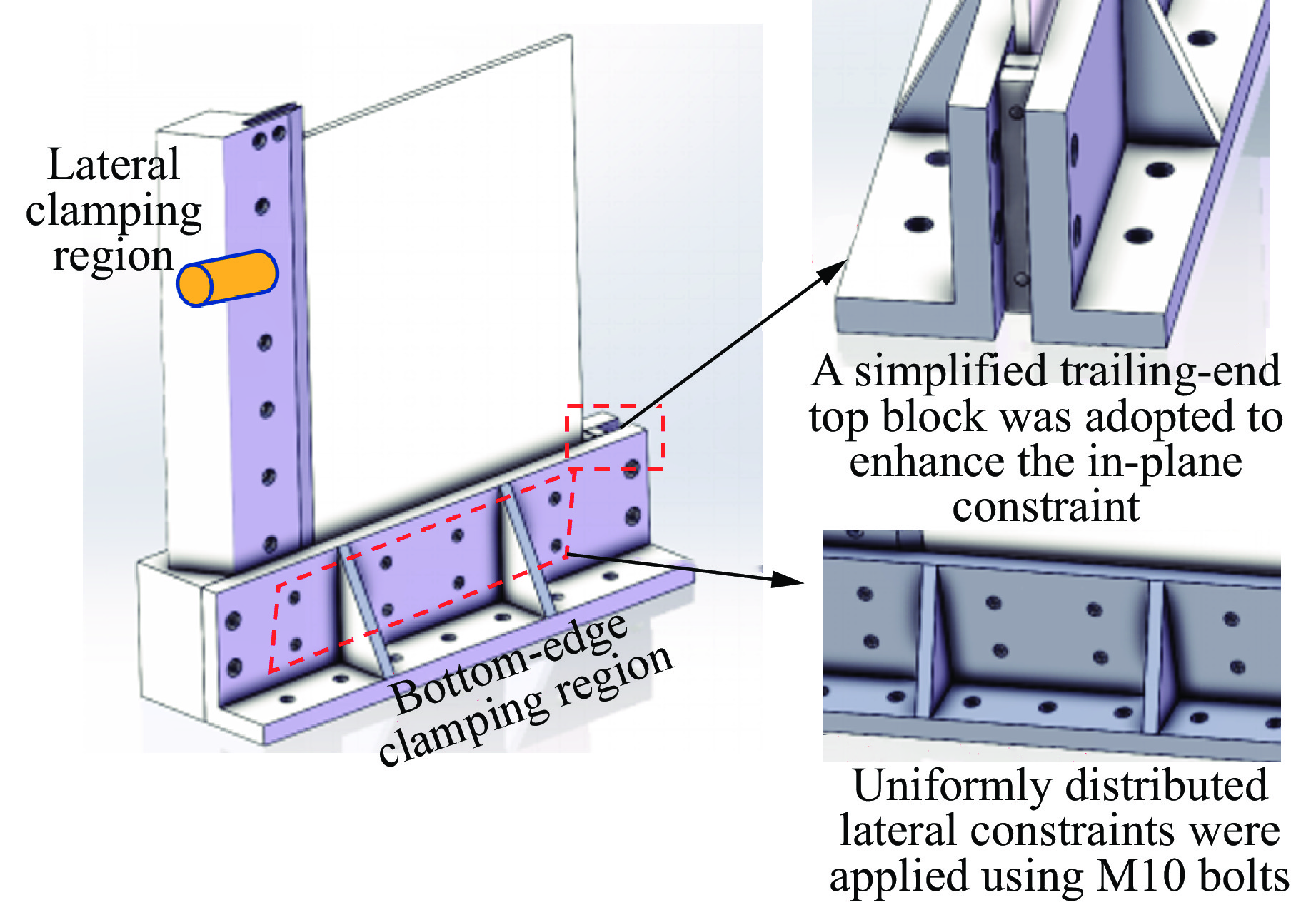
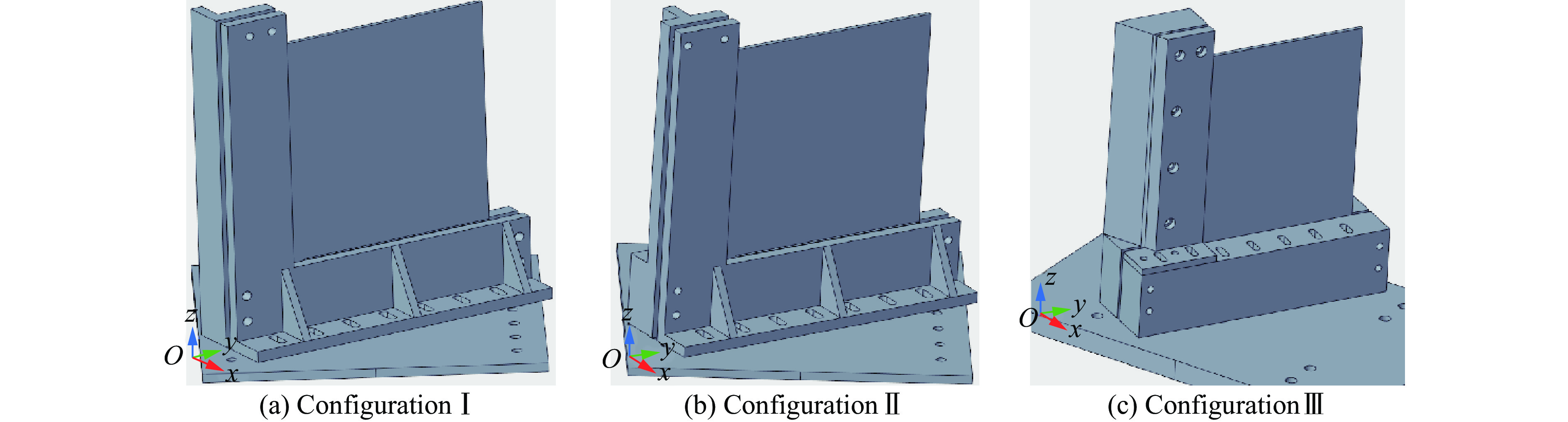
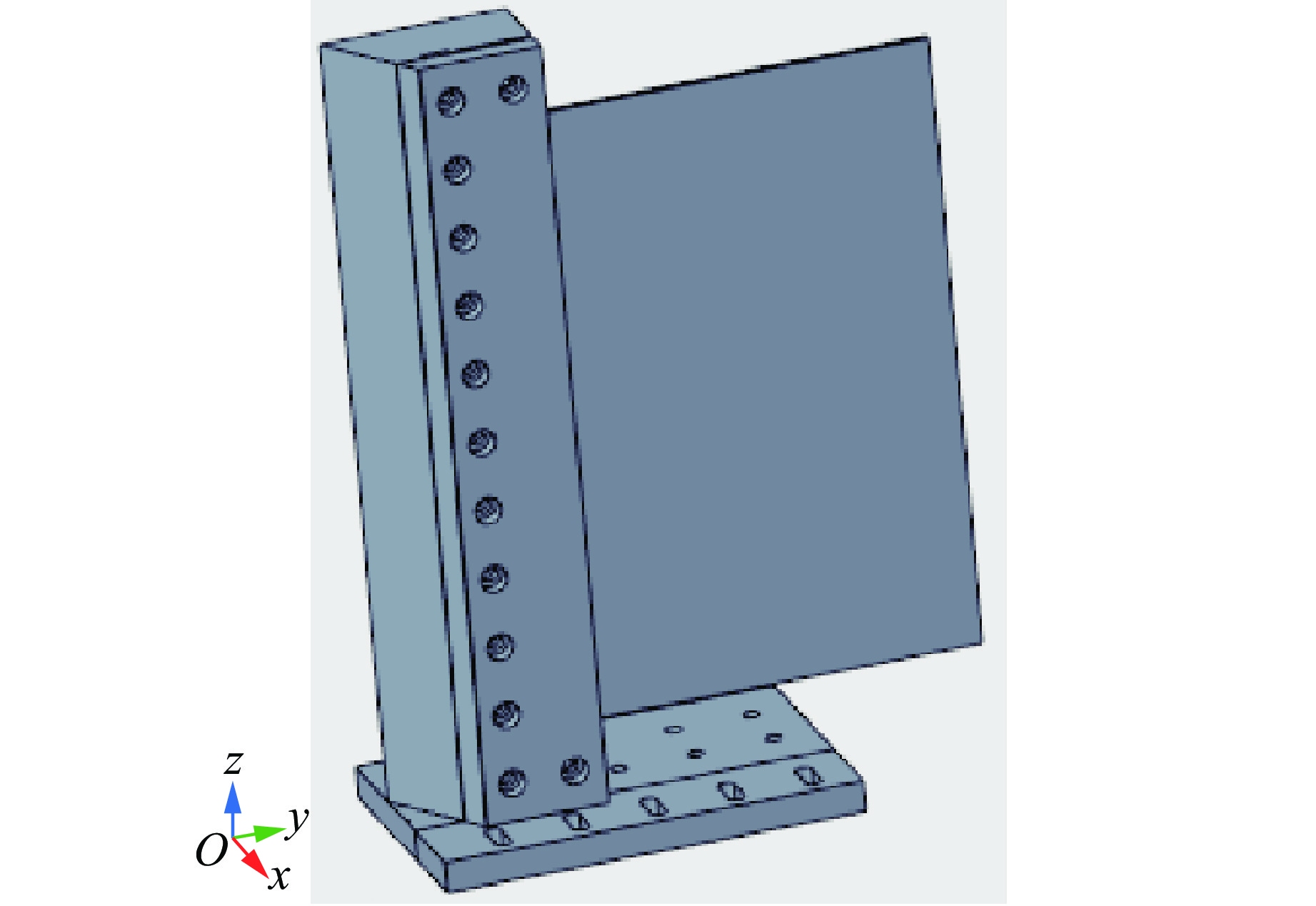
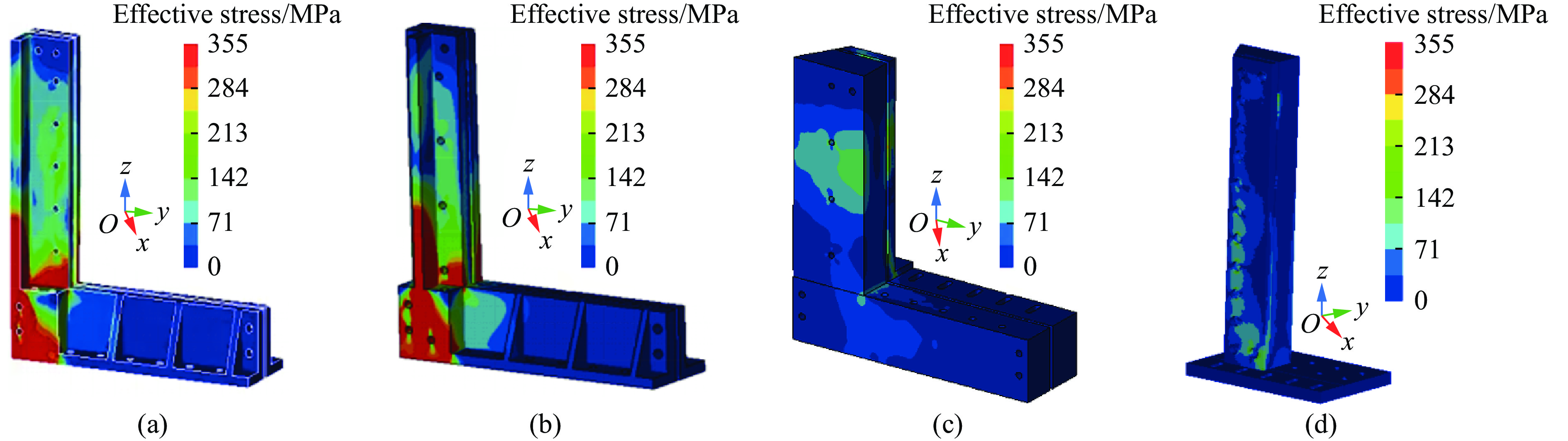
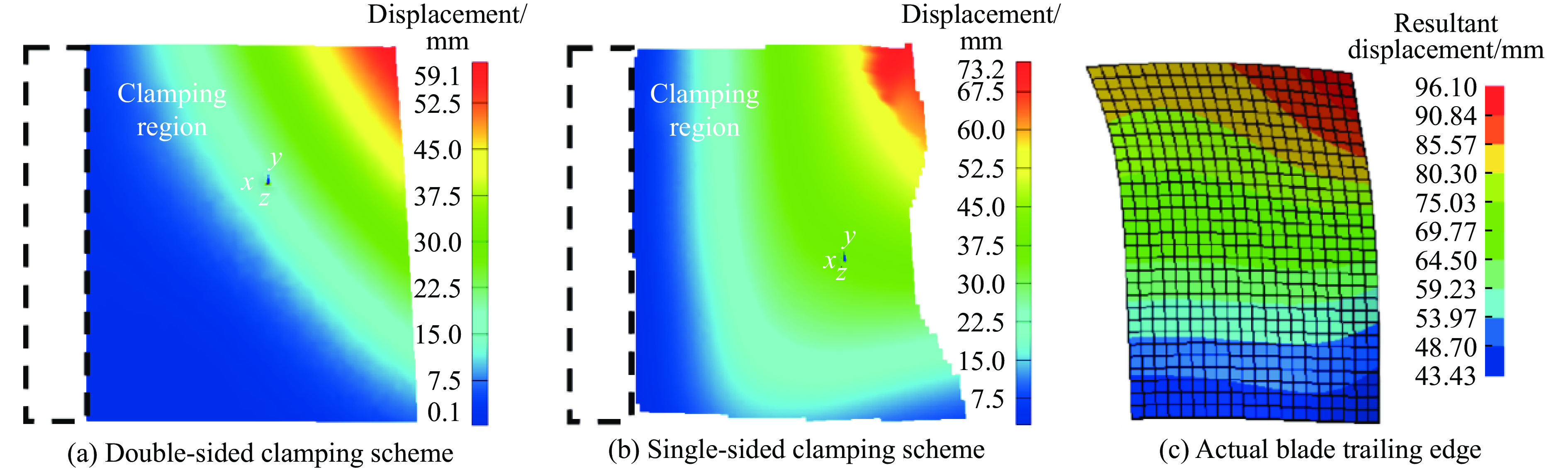
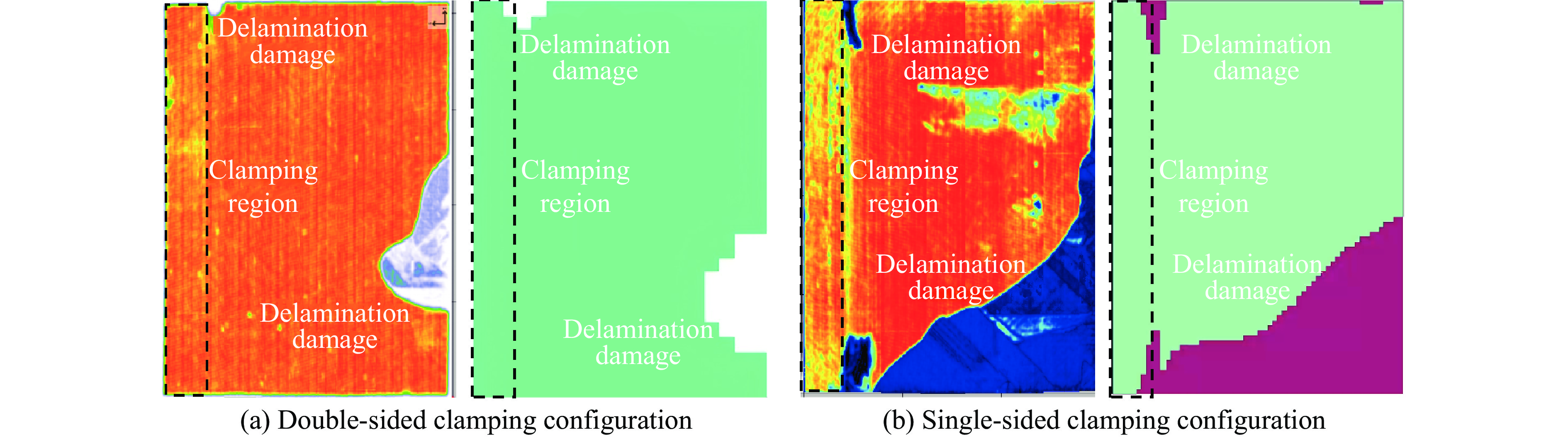
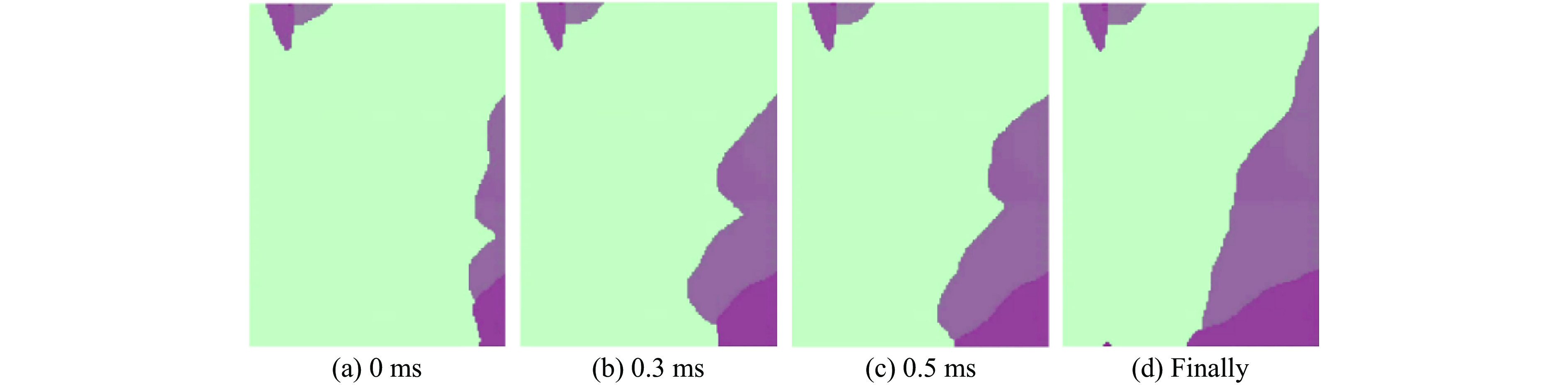
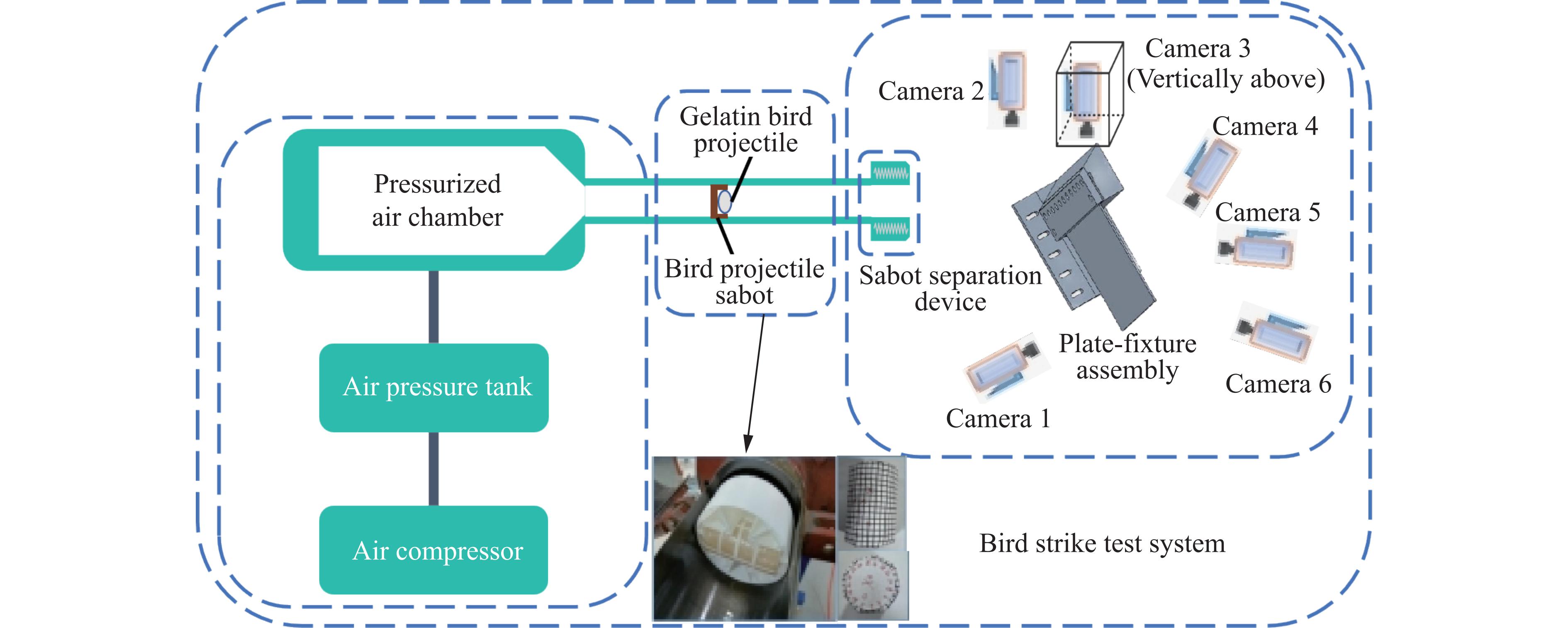
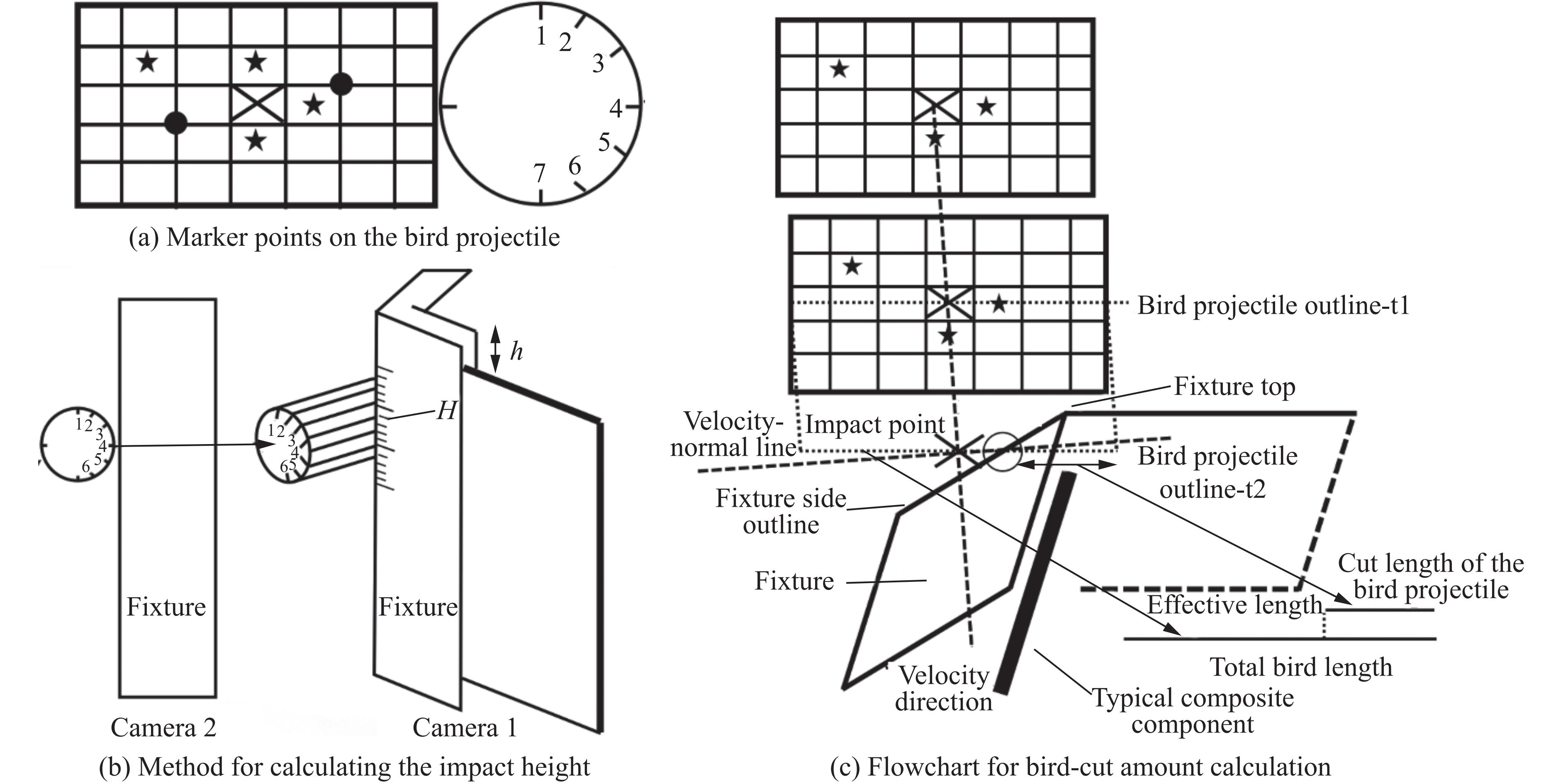
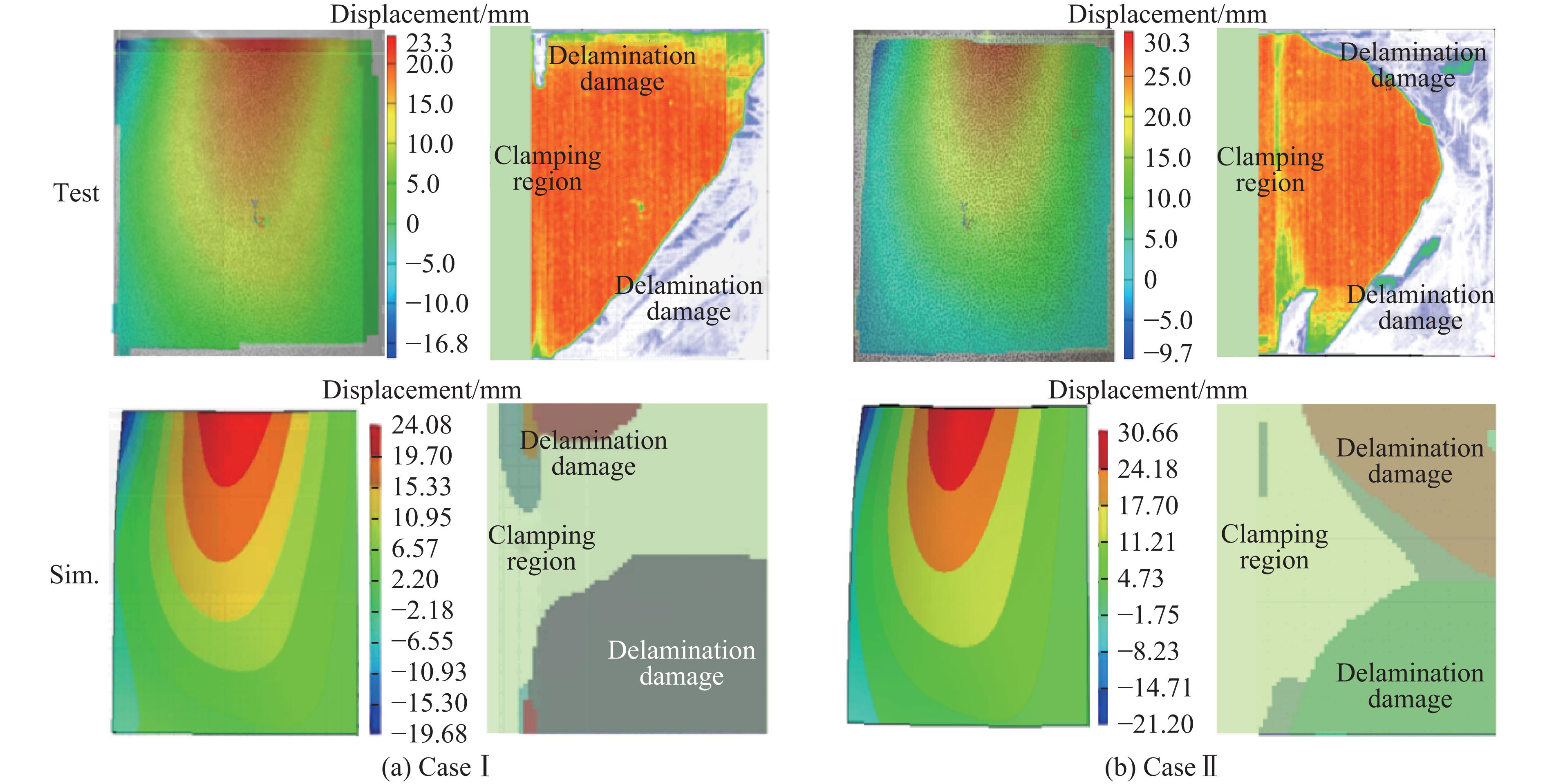
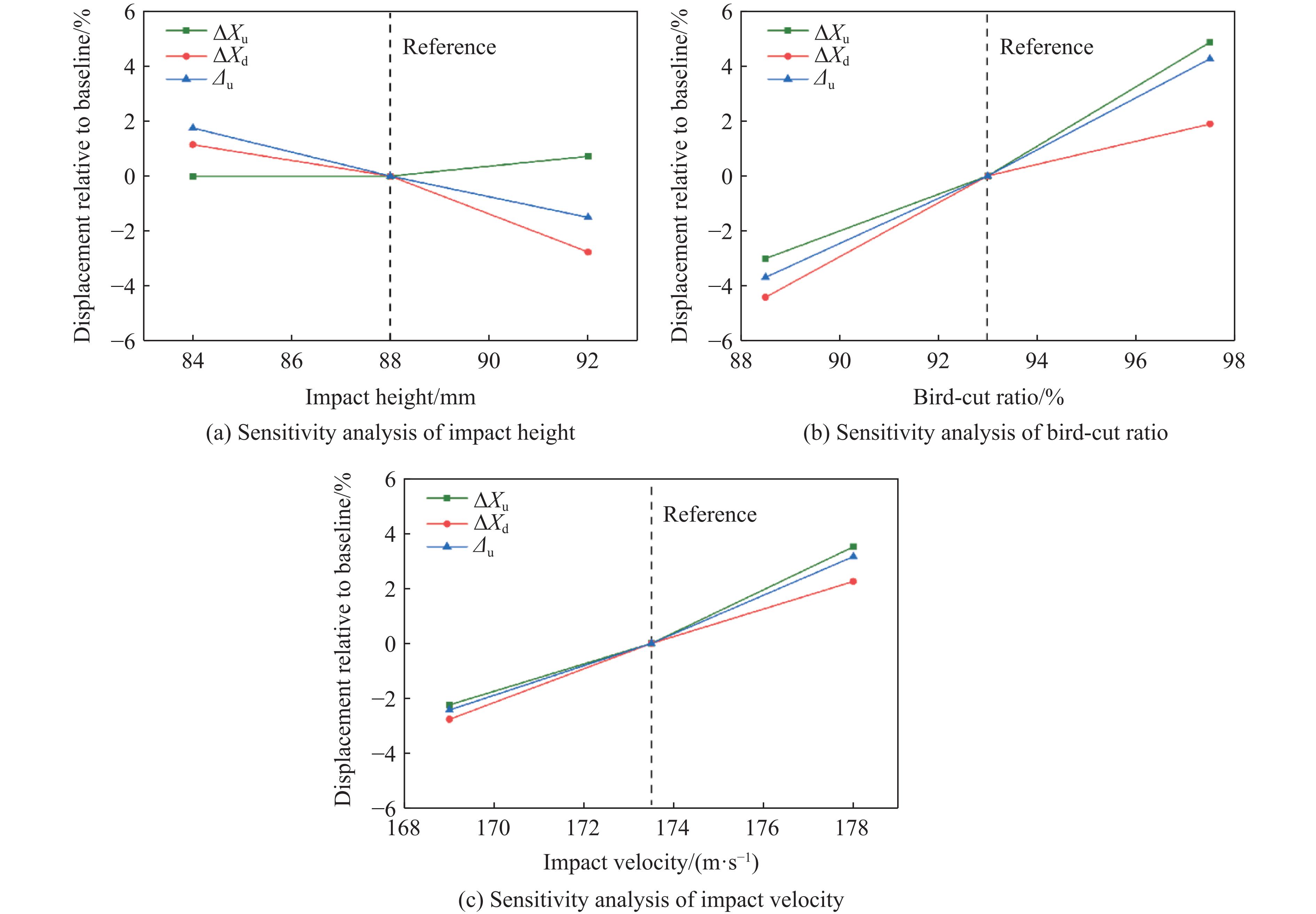
To investigate the response and damage behavior of composite aero-engine blades under bird-strike events, an equivalent bird-strike testing method was proposed in which a component-level flat plate specimen was used to replace a full-scale fan blade. The method aims to reproduce the trailing-edge delamination damage observed in full-scale blades during bird strike. Bird-strike tests and corresponding numerical simulations on the plate specimens under different clamping configurations were conducted, and the impact response characteristics as well as the initiation and propagation processes of delamination were systematically analyzed for each configuration. Based on these results, a component-level equivalent test methodology capable of effectively simulating trailing-edge delamination in blade bird-strike scenarios is proposed. A baseline impact condition that induces single-side trailing-edge delamination in a representative composite laminate is identified, including impact height, impact velocity, and the bird-cut ratio (defined as the percentage of the effective impacting volume of the bird projectile relative to its total volume at the instant of impact). In addition, by comparing test and numerical results under various impact conditions, the accuracy of the numerical model is validated. Using the experimentally validated model, sensitivity analyses were performed with respect to the test parameters (impact height, impact velocity, and bird-cut ratio). The results show that, within the controllable ranges of parameter variation in the tests, the changes in key impact response metrics of the composite plate—namely the peak displacement at the upper trailing edge, the peak displacement at the lower trailing edge, and the displacement difference along the upper edge—are all less than 5% relative to the baseline condition. This study demonstrates that the proposed equivalent testing method enables a composite plate test to replicate the local displacement response and delamination pattern of a full-scale blade under bird strike, and the test outcomes exhibit good robustness.

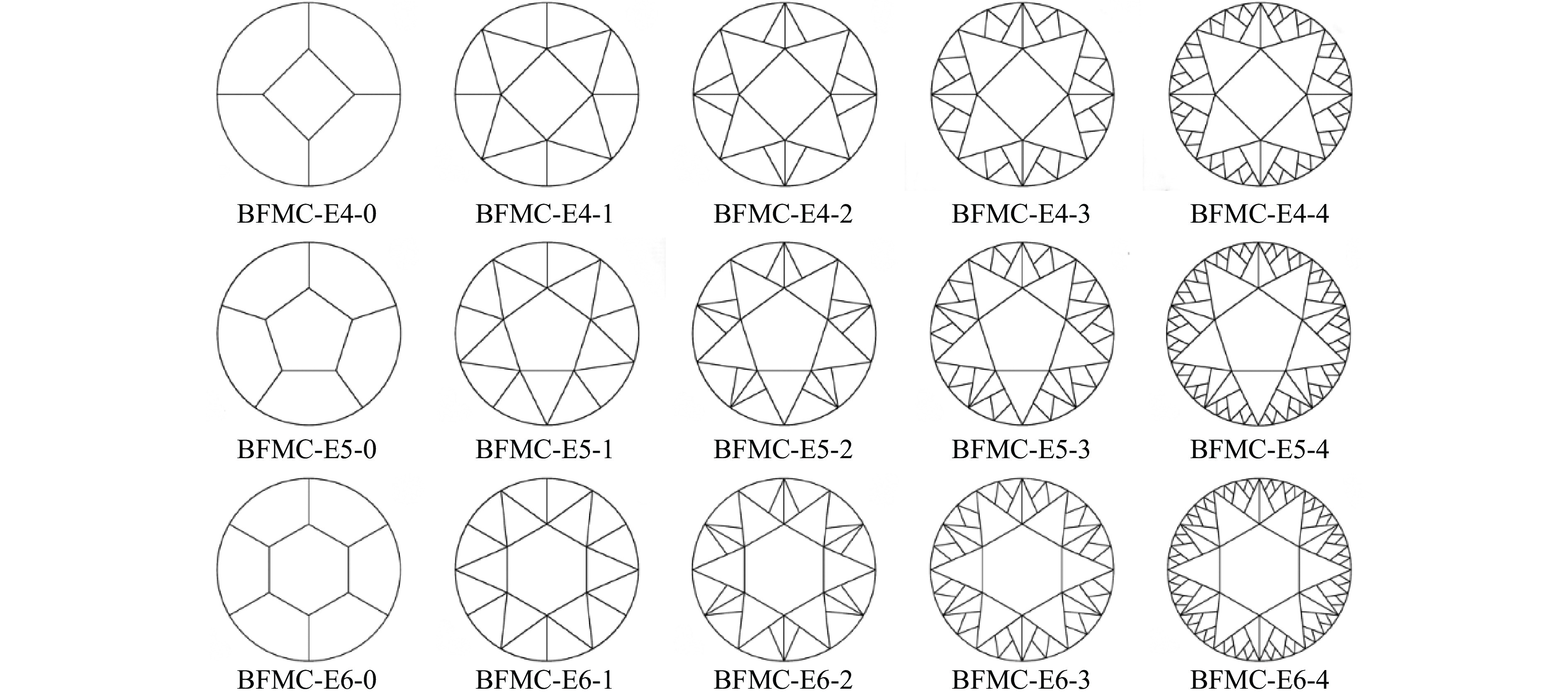
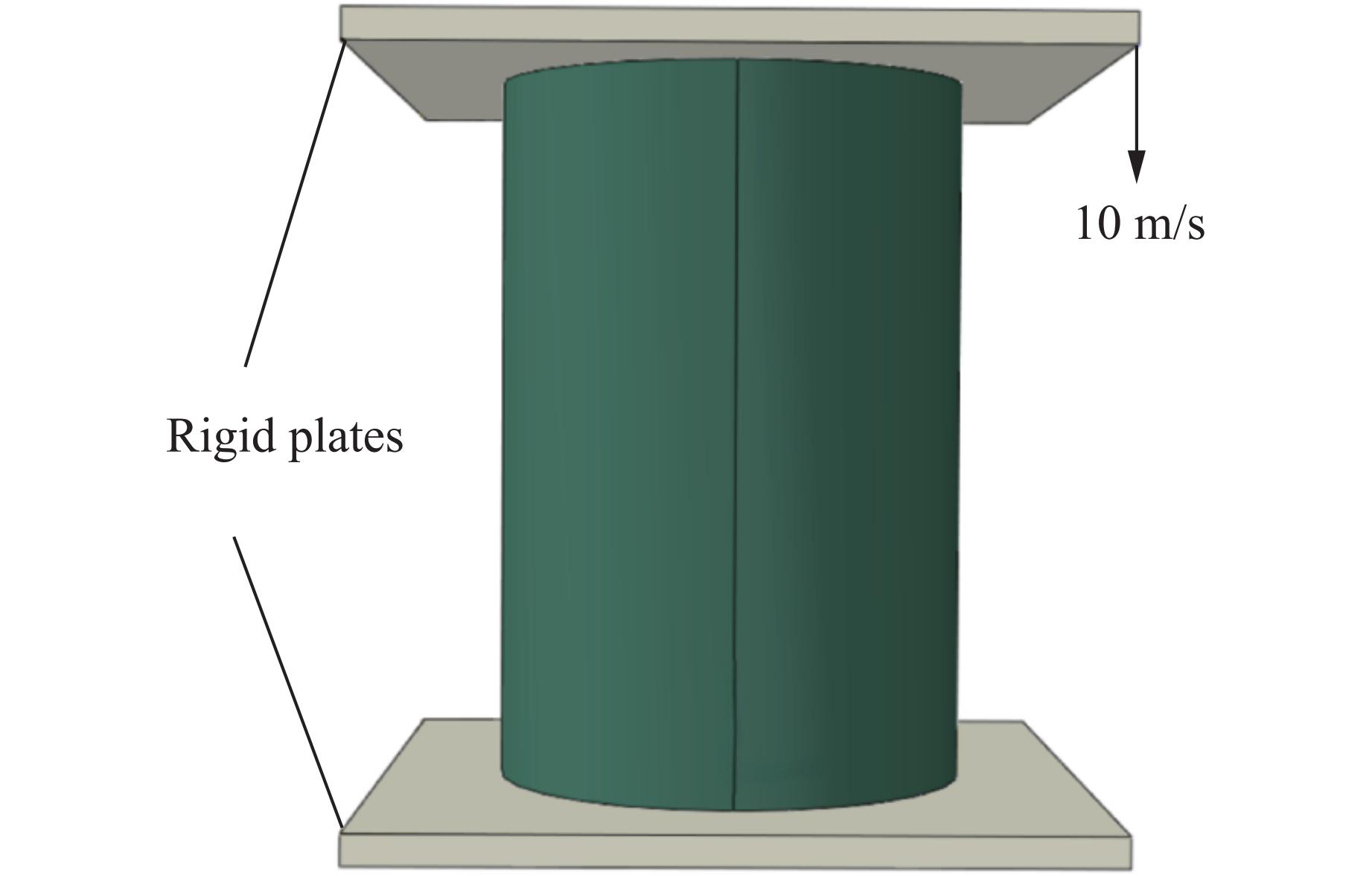
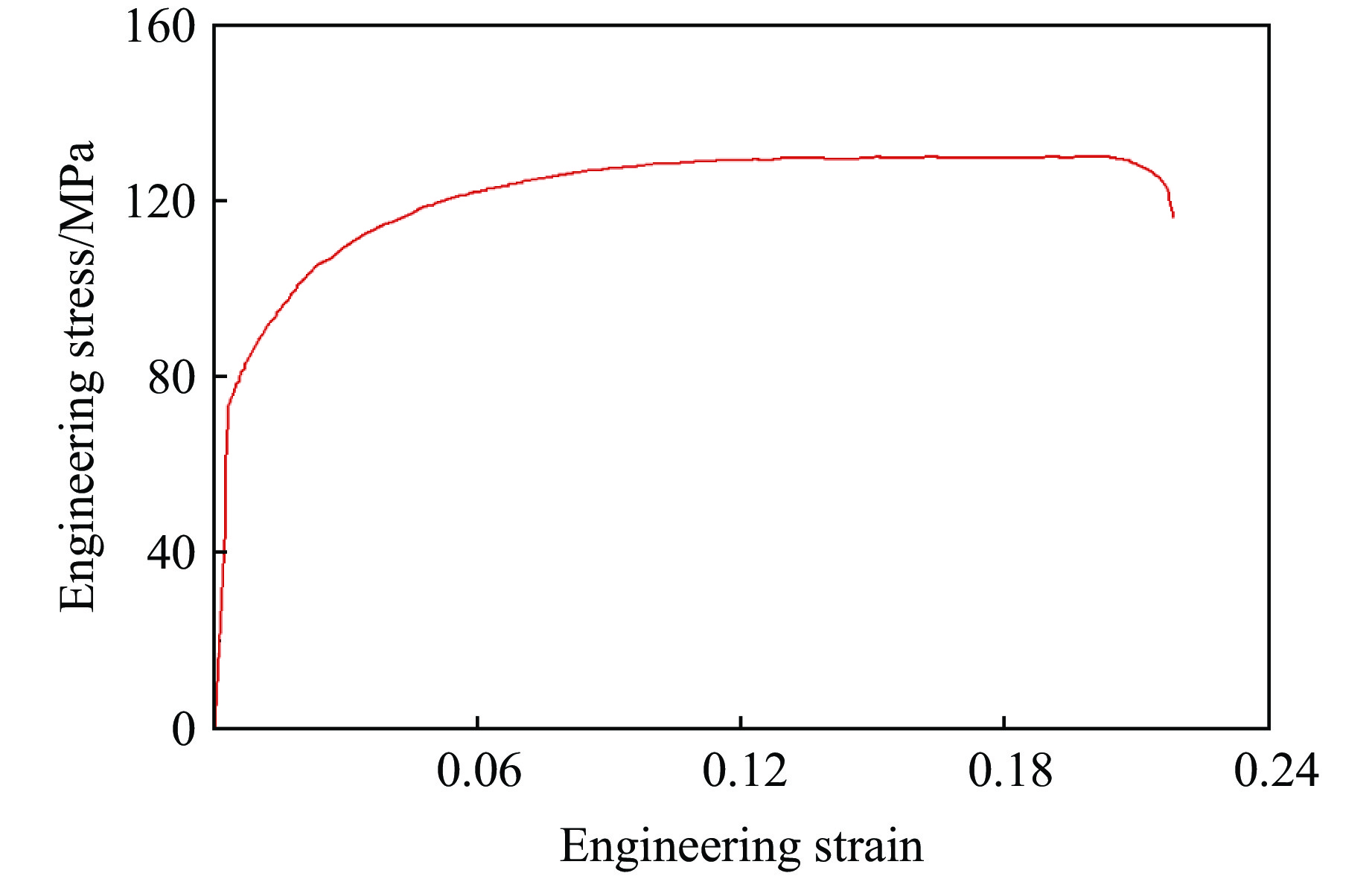
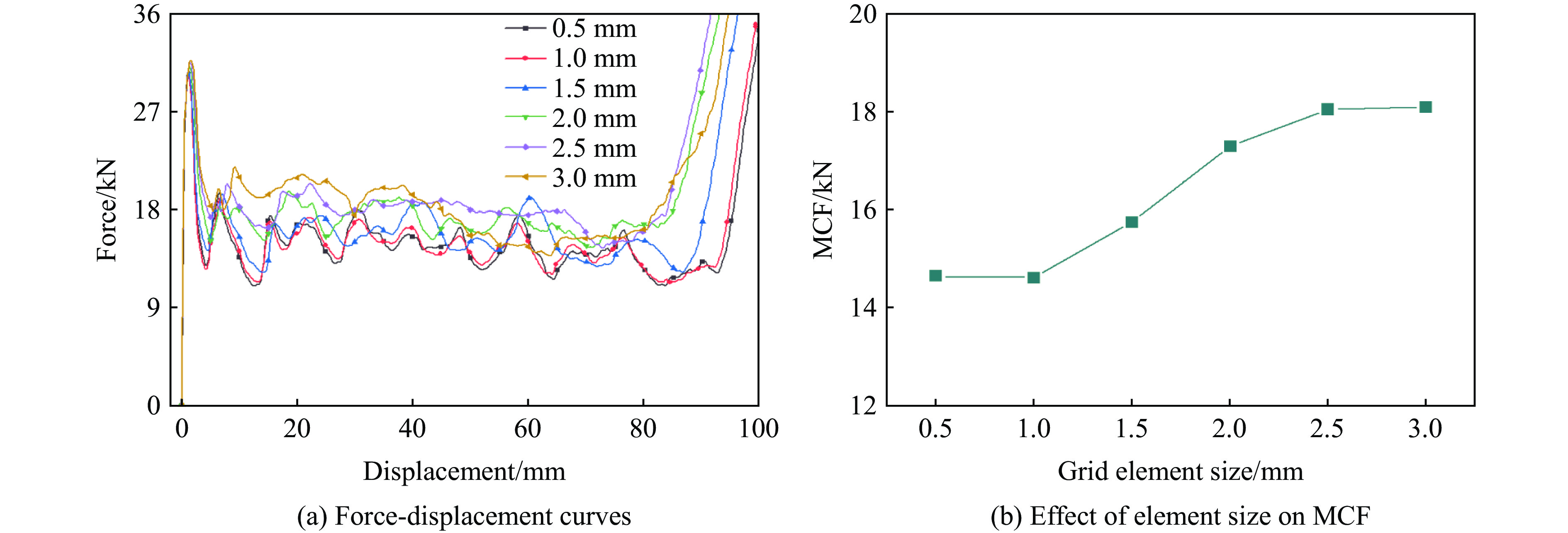
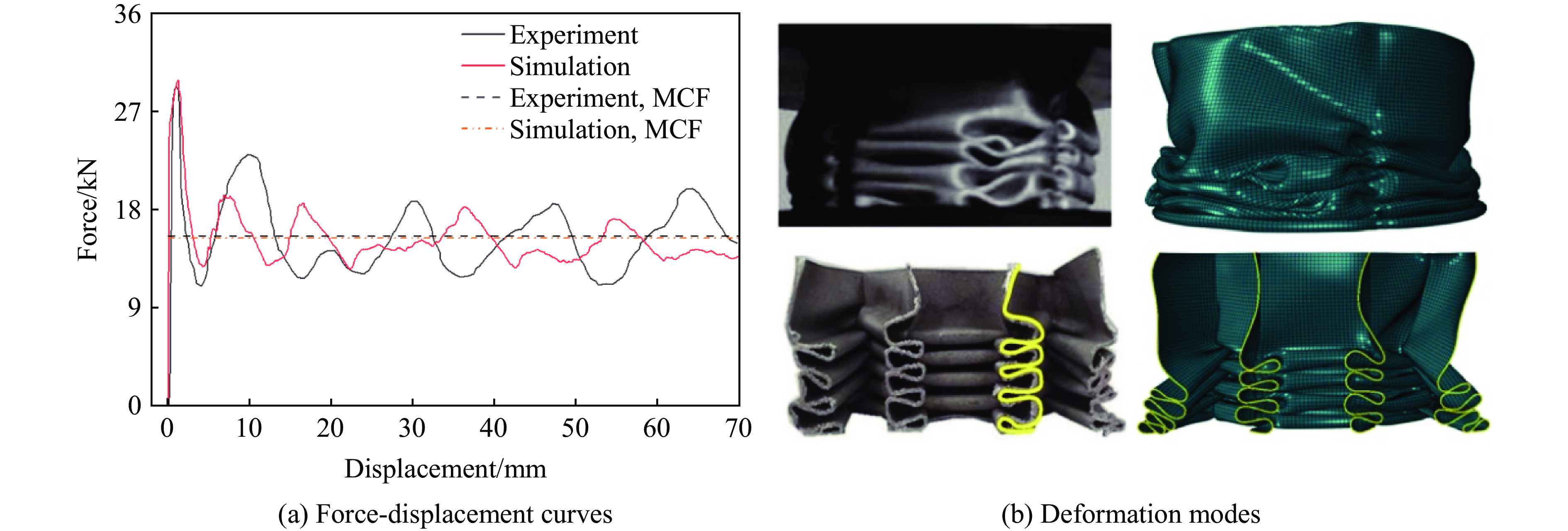
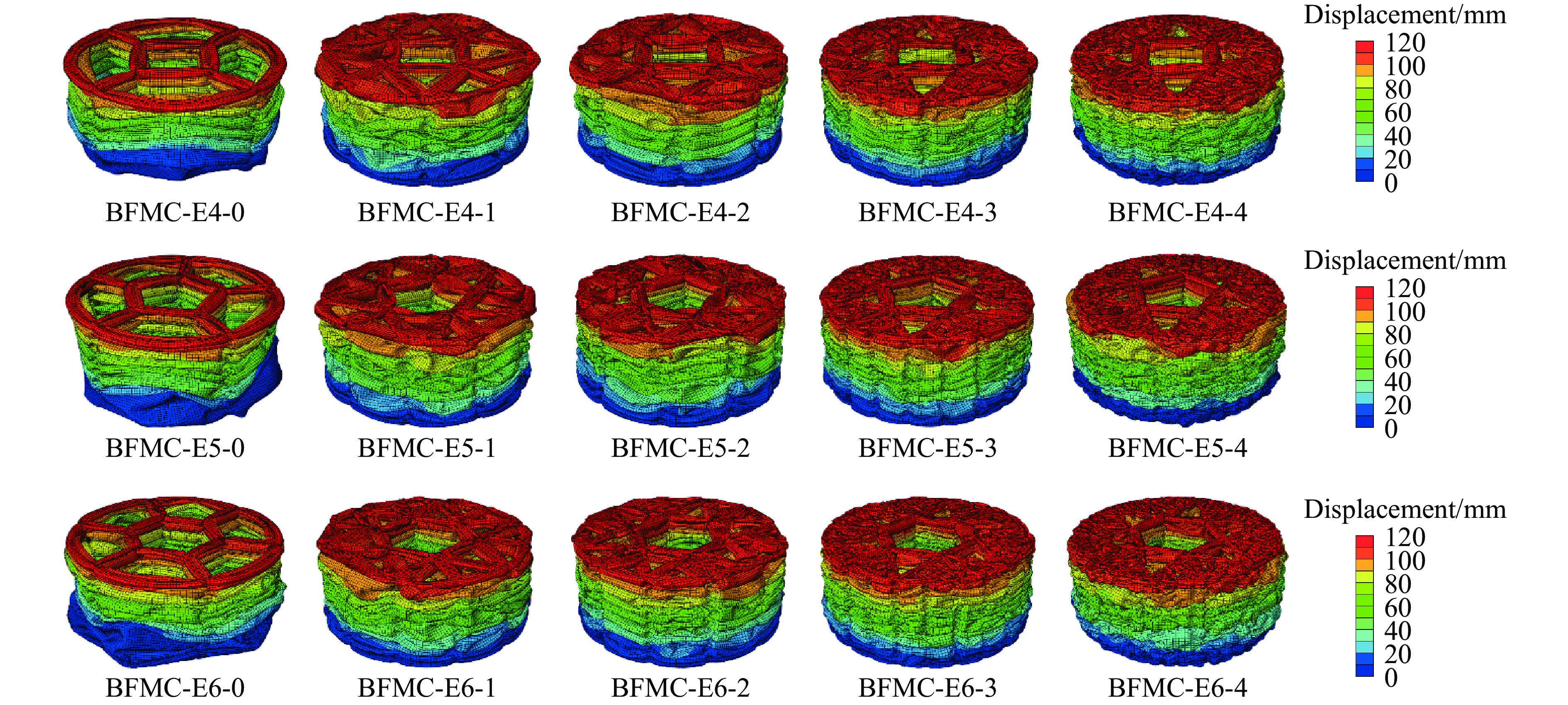
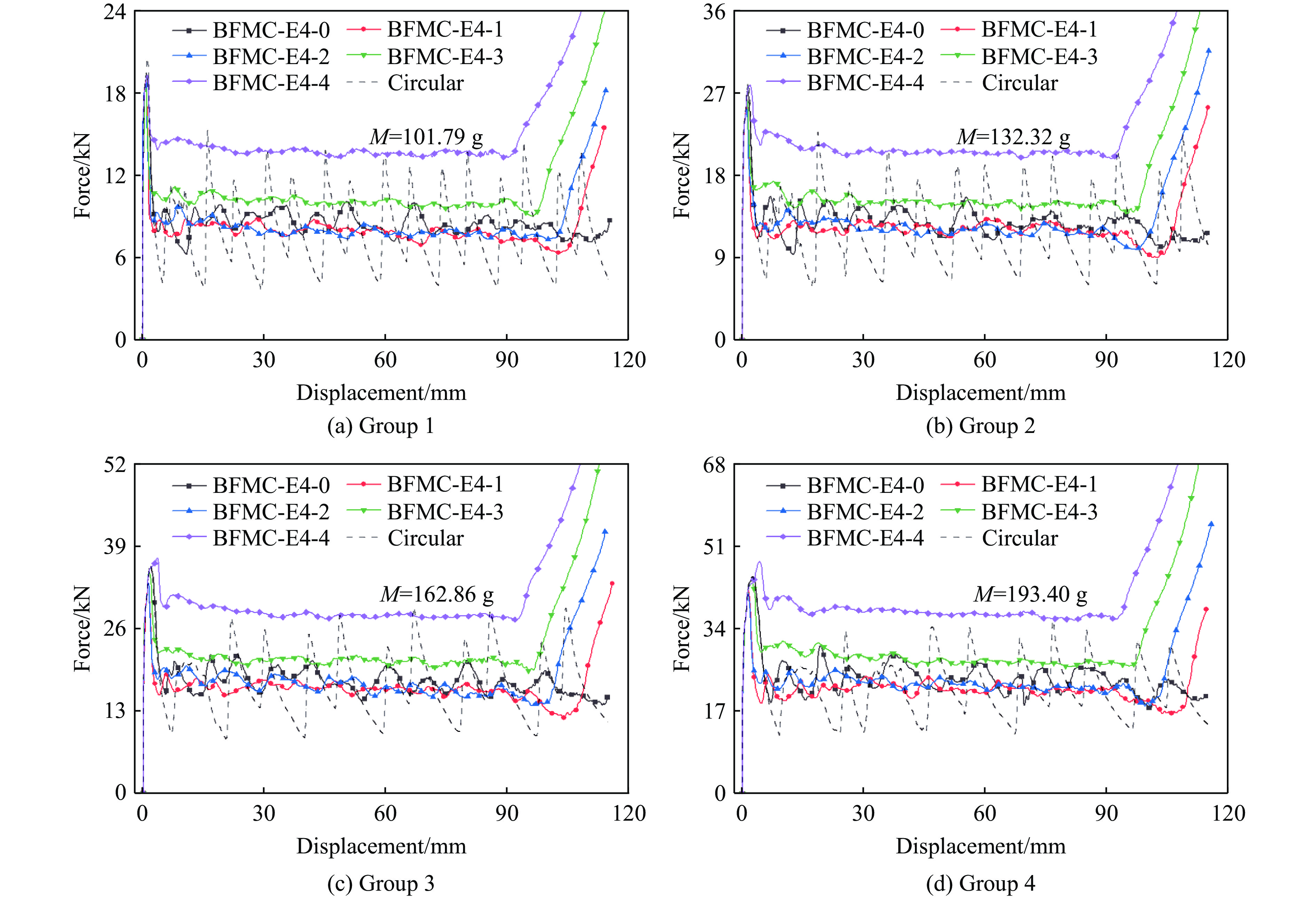
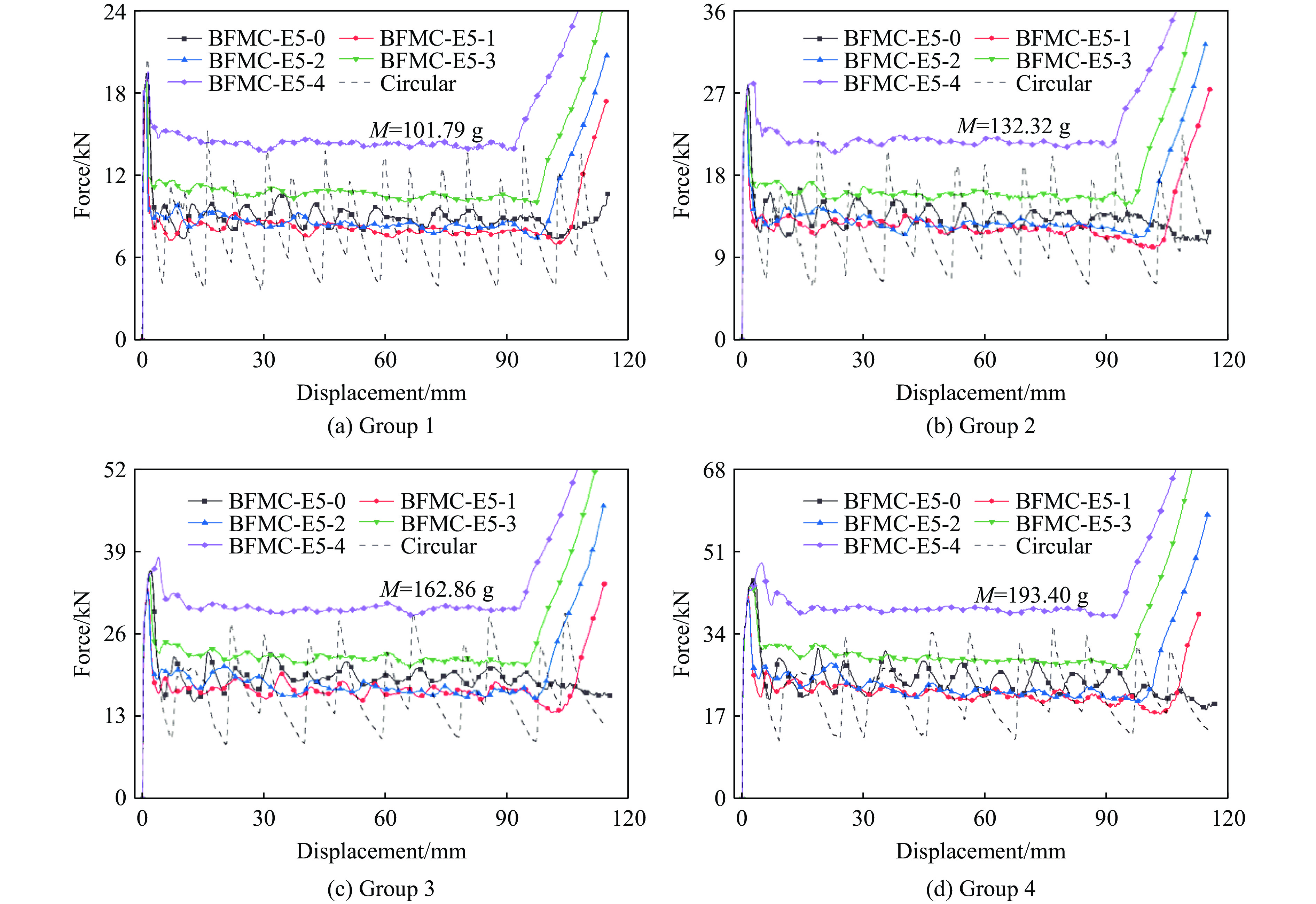
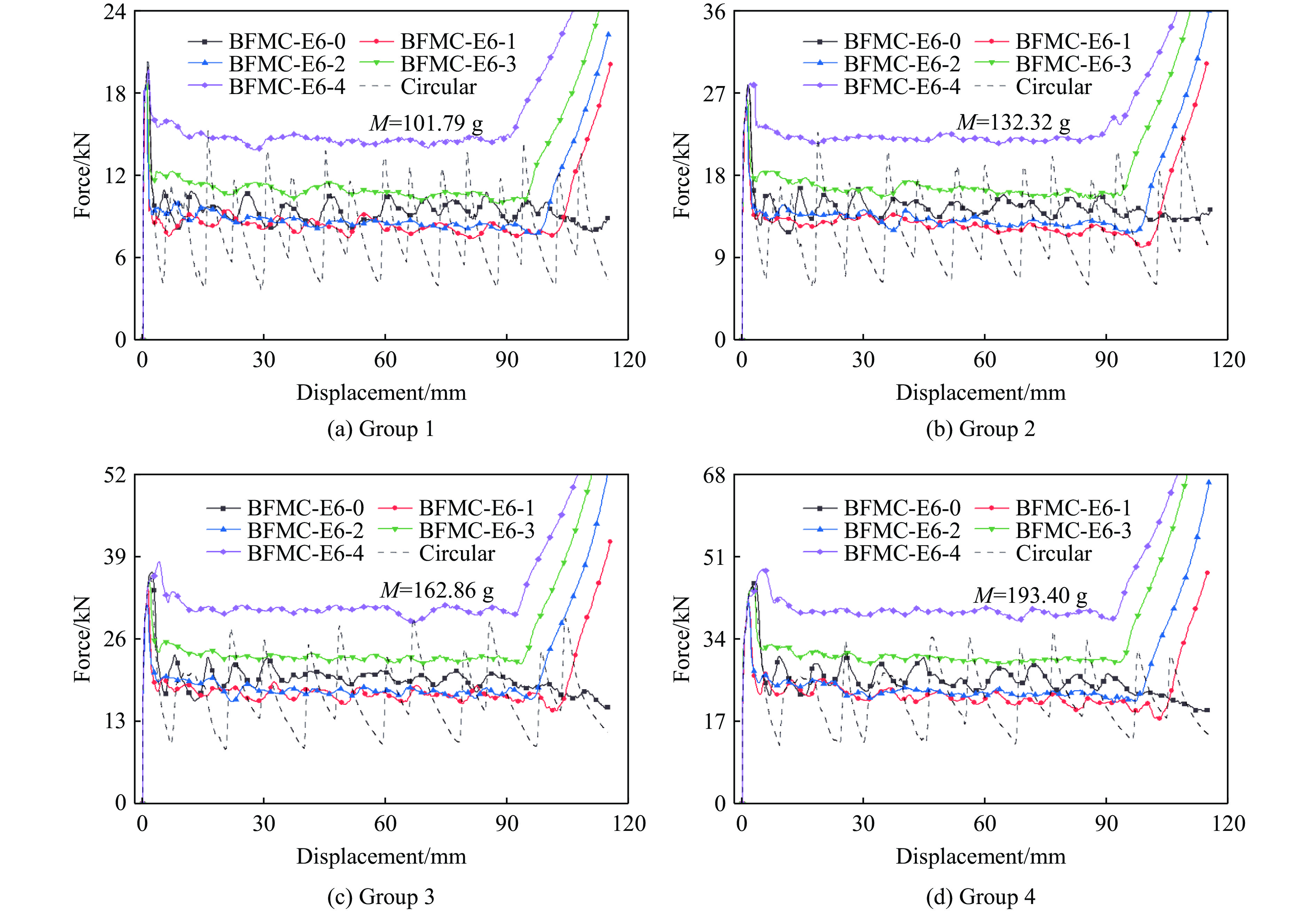
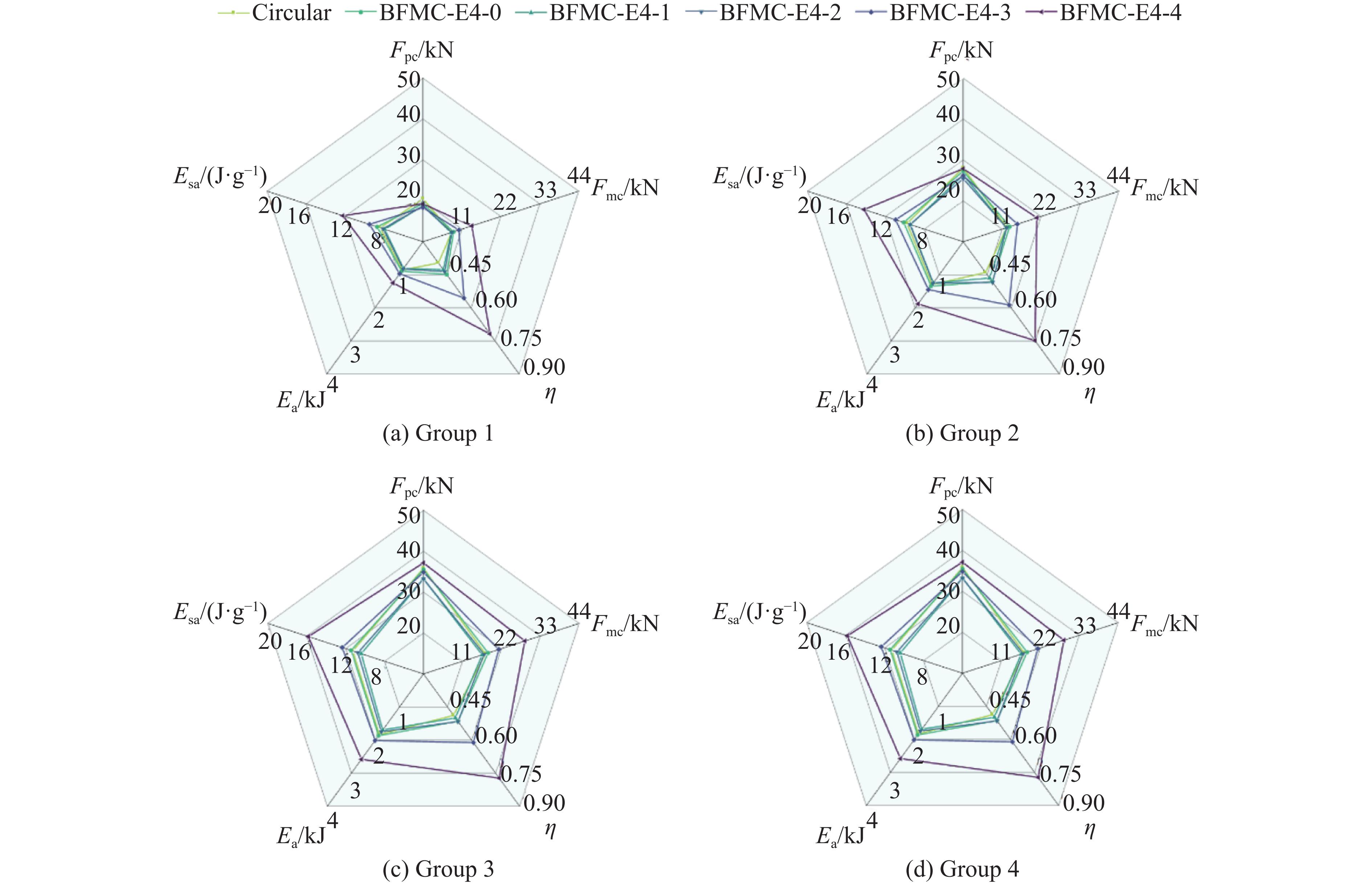
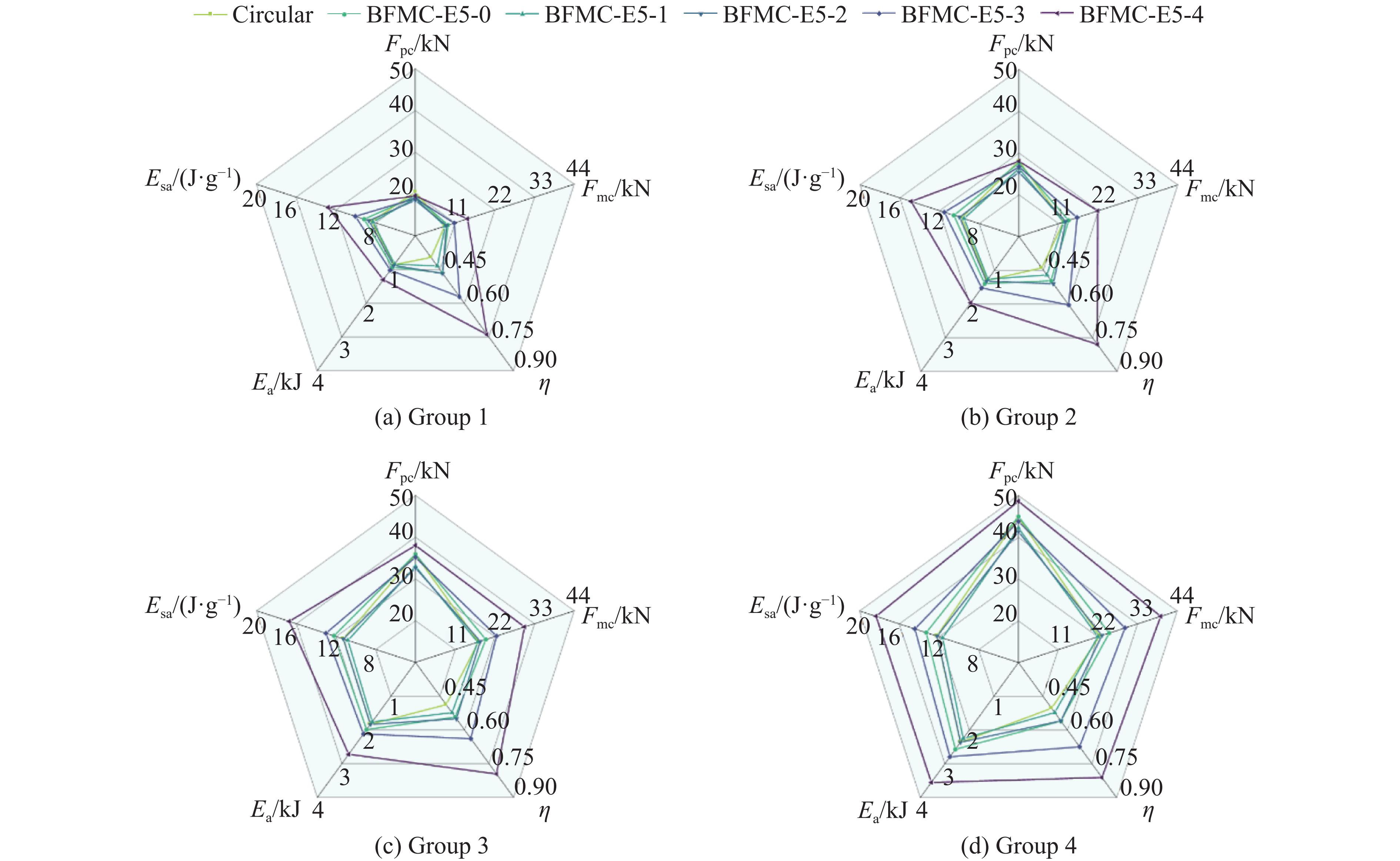
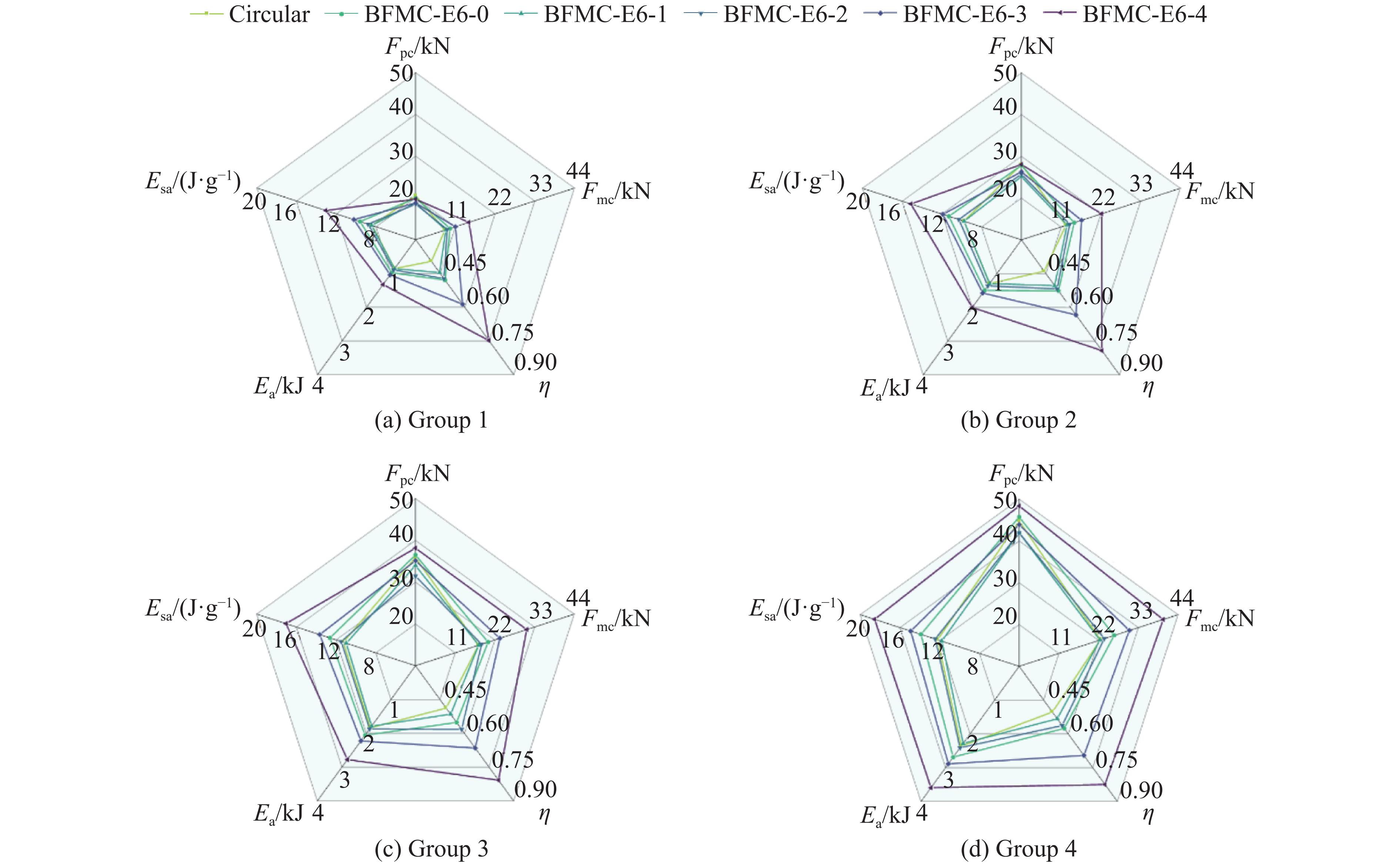
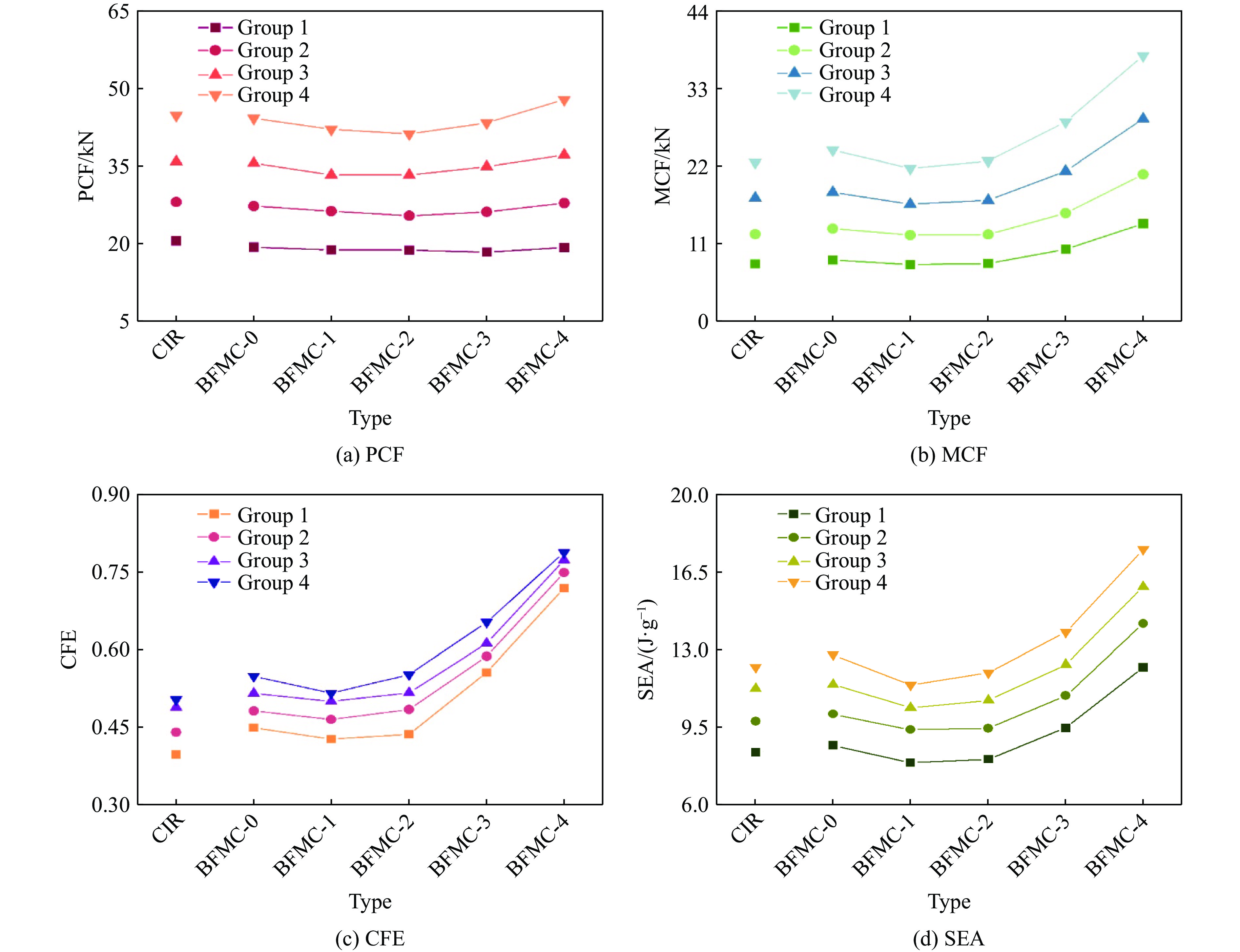
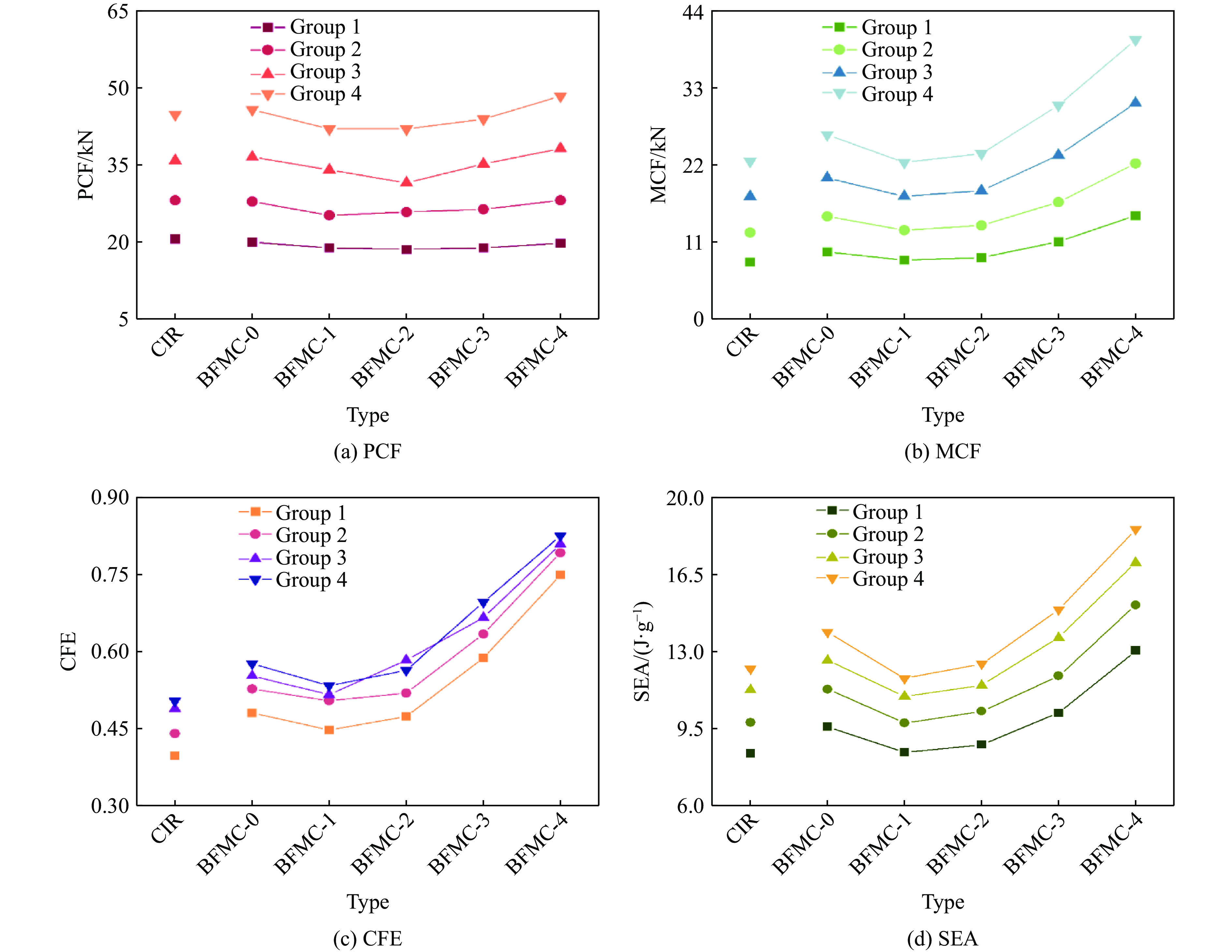
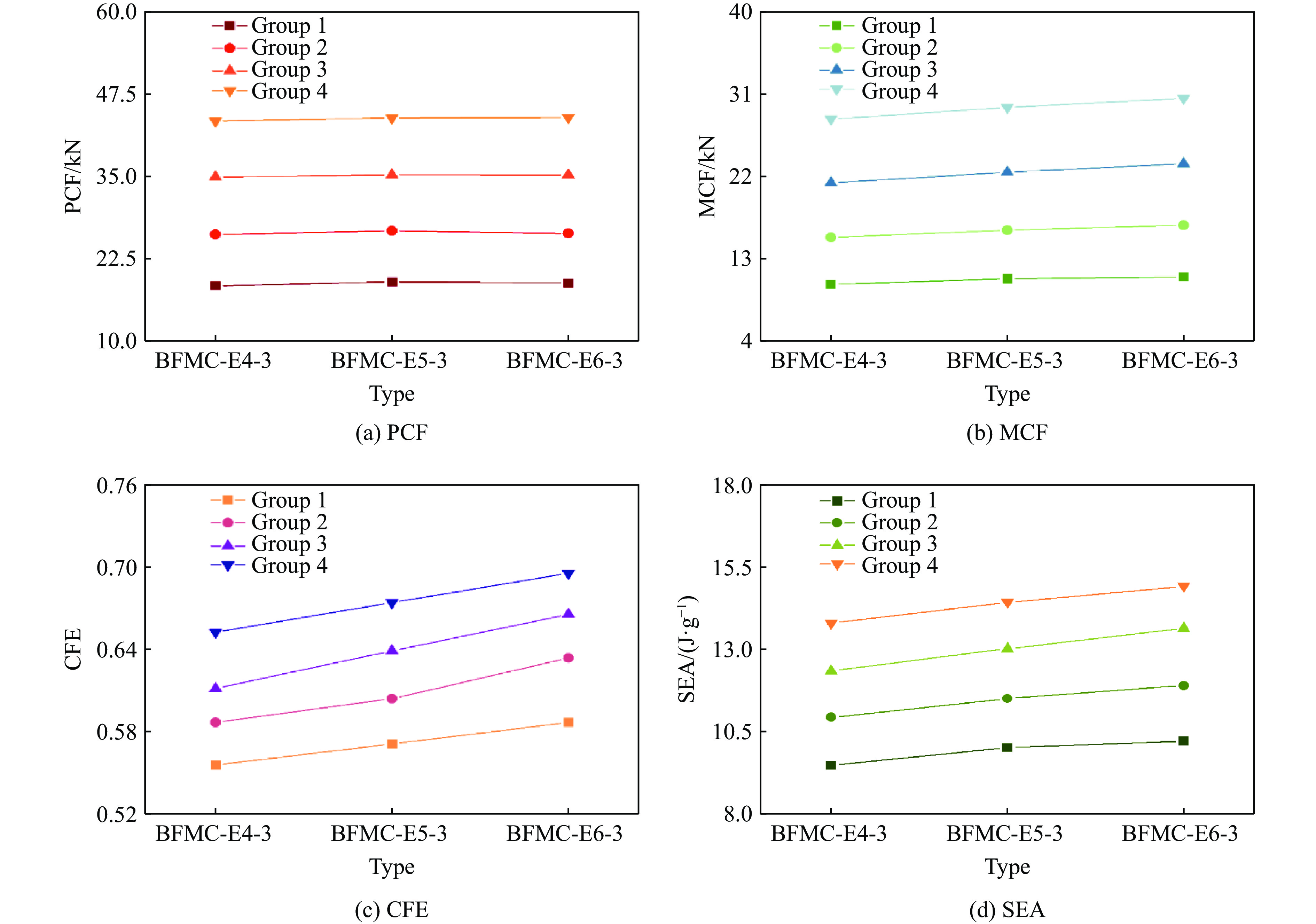
A bio-inspired fractal multi-cell circular (BFMC) tube with embedded regular polygons is proposed to address the gap between the need for high absorption and the limited performance of traditional thin-walled circular tubes. Inspired by biological structures and fractal hierarchy theory, geometric models of BFMC tubes embedded with square, pentagonal, and hexagonal cells are constructed. Numerical simulations are carried out to systematically investigate the effects of mass, fractal dimension, and the number of sides of the embedded polygons on the axial crushing performance, and the results are compared with those of typical multi-tubes. The results indicate that, under approximately equal mass conditions, the BFMC tube can significantly enhance the specific energy absorption and the load-bearing capacity owing to its fractal hierarchical and bio-inspired configurations. Its crashworthiness increases with mass, first decreases and then rises as the fractal dimension increases, and improves further as the number of polygon sides increases, while the peak force is only weakly affected. A theoretical model for predicting the mean crushing force of BFMC tubes is developed based on the super folding element theory and is validated through numerical simulations. This study provides theoretical support and structural design guidelines for developing high performance thin-walled energy absorption structures.





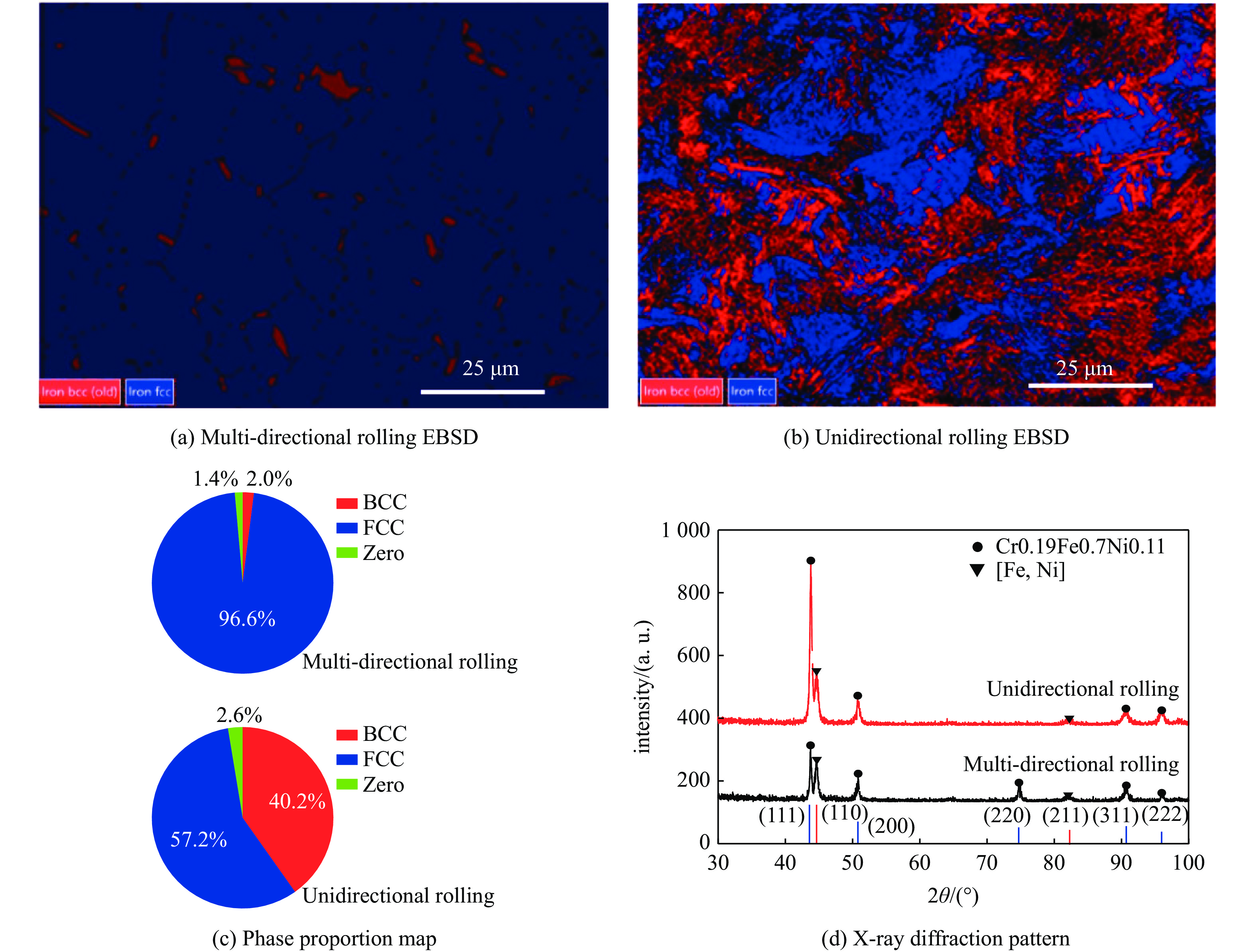
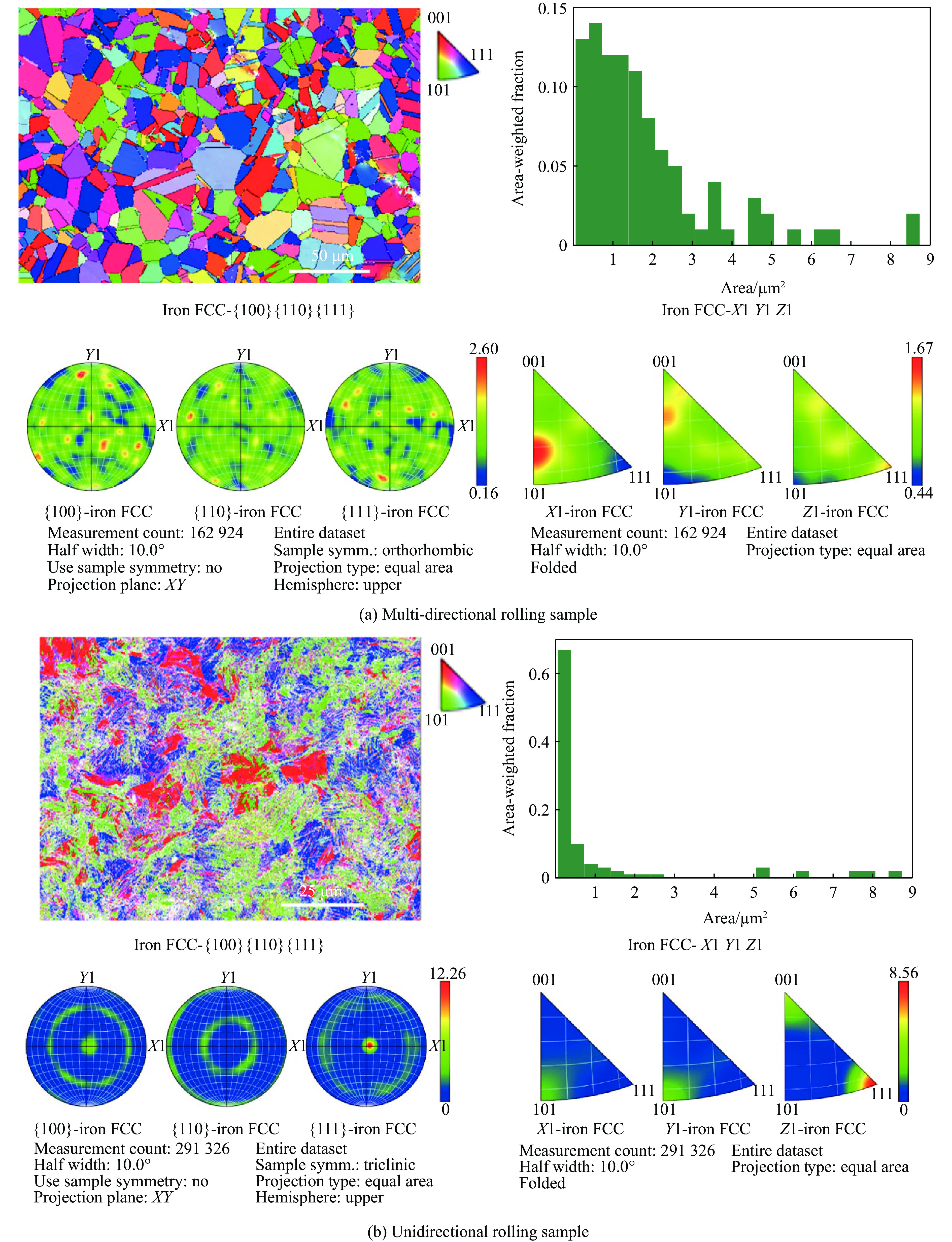
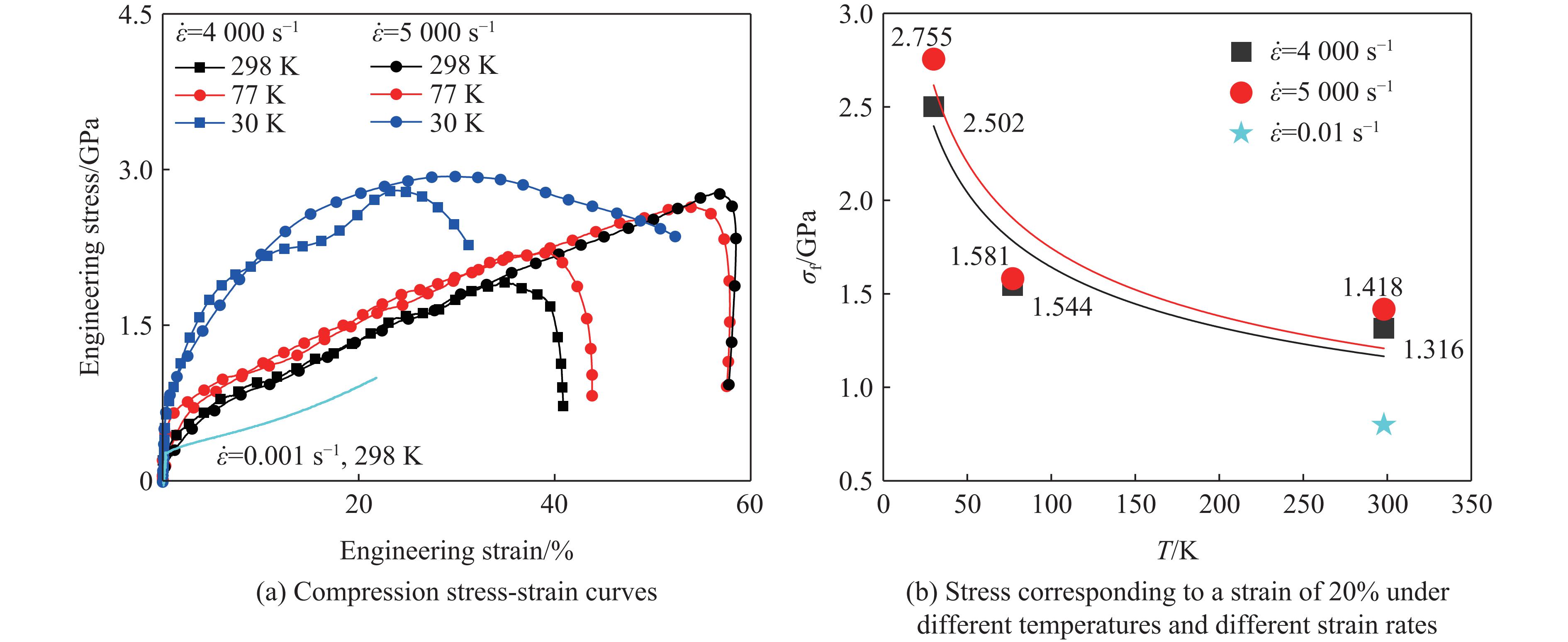
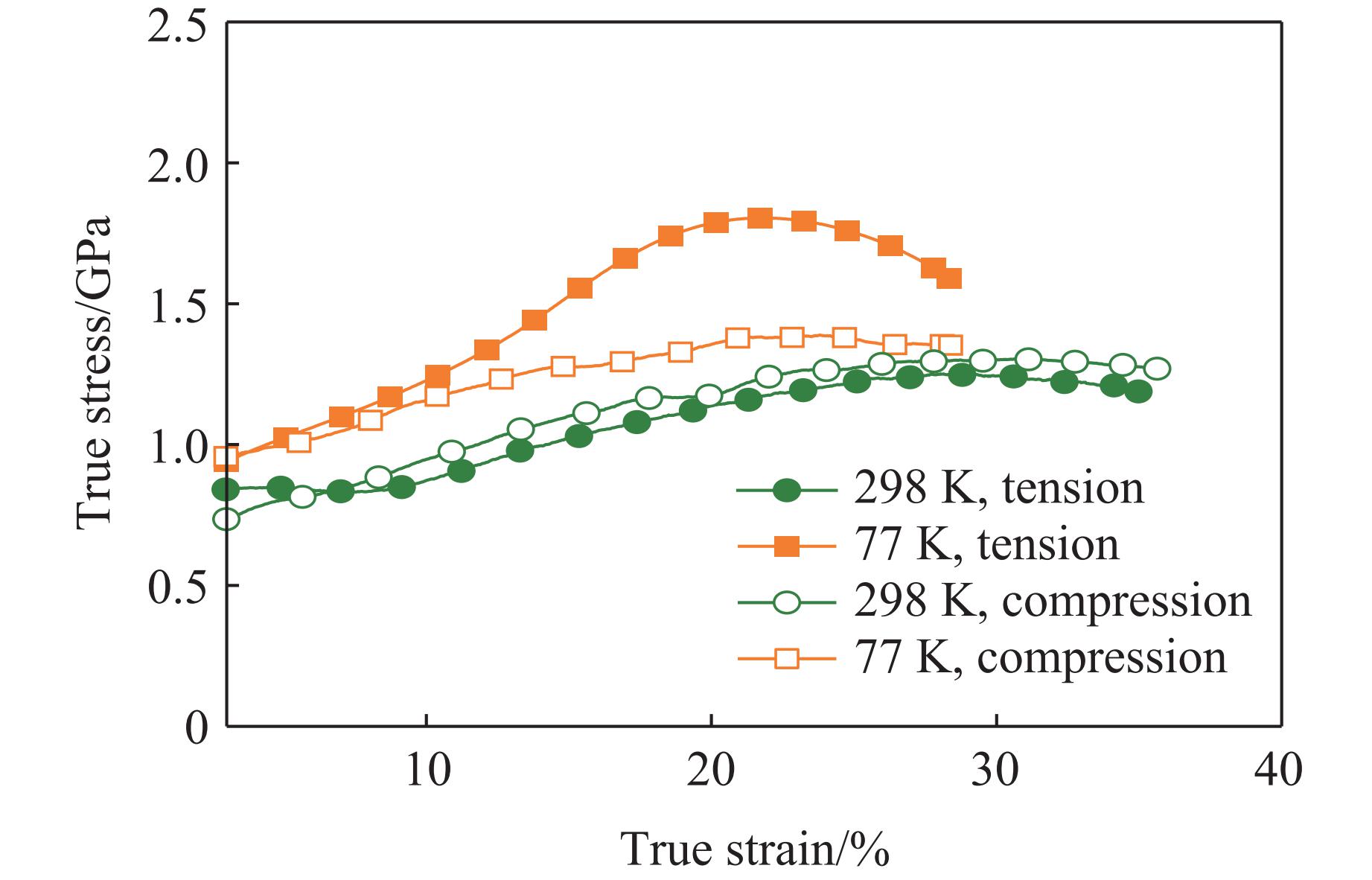
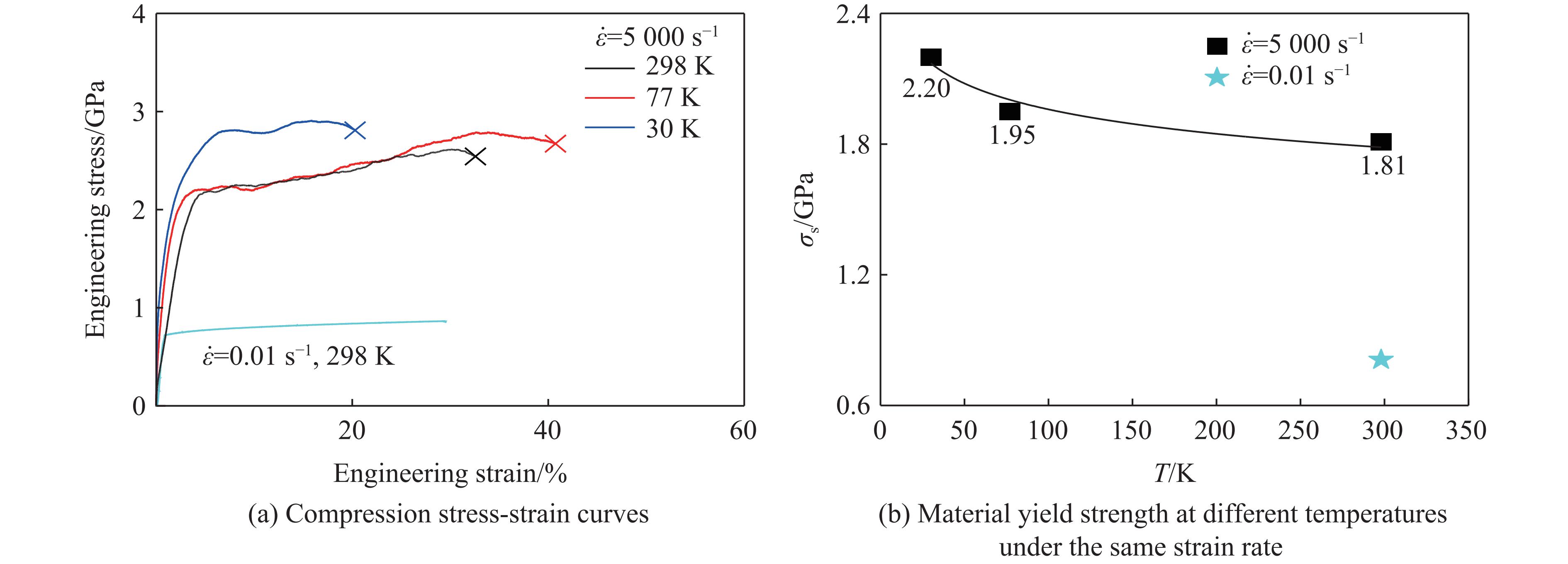
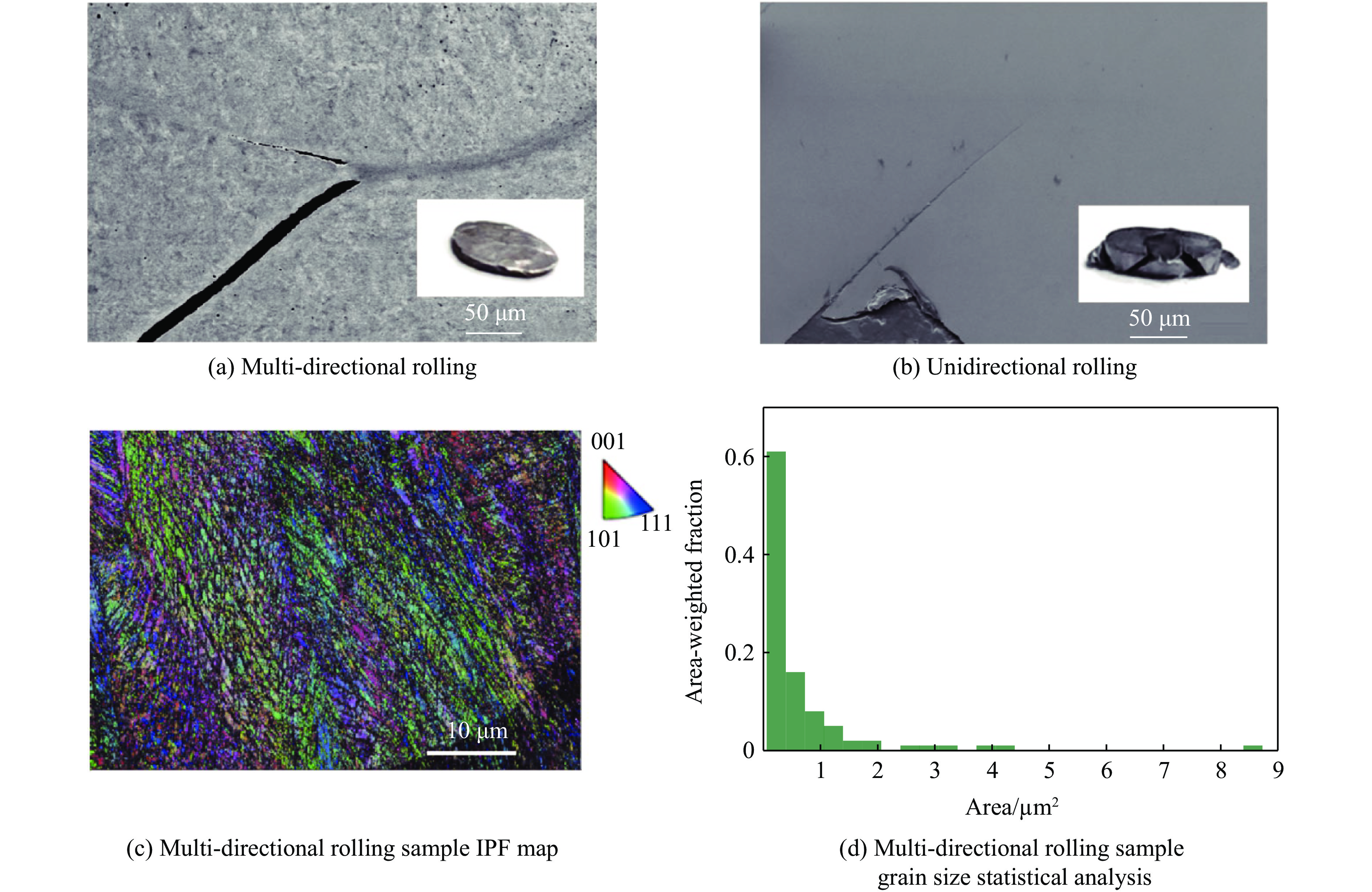
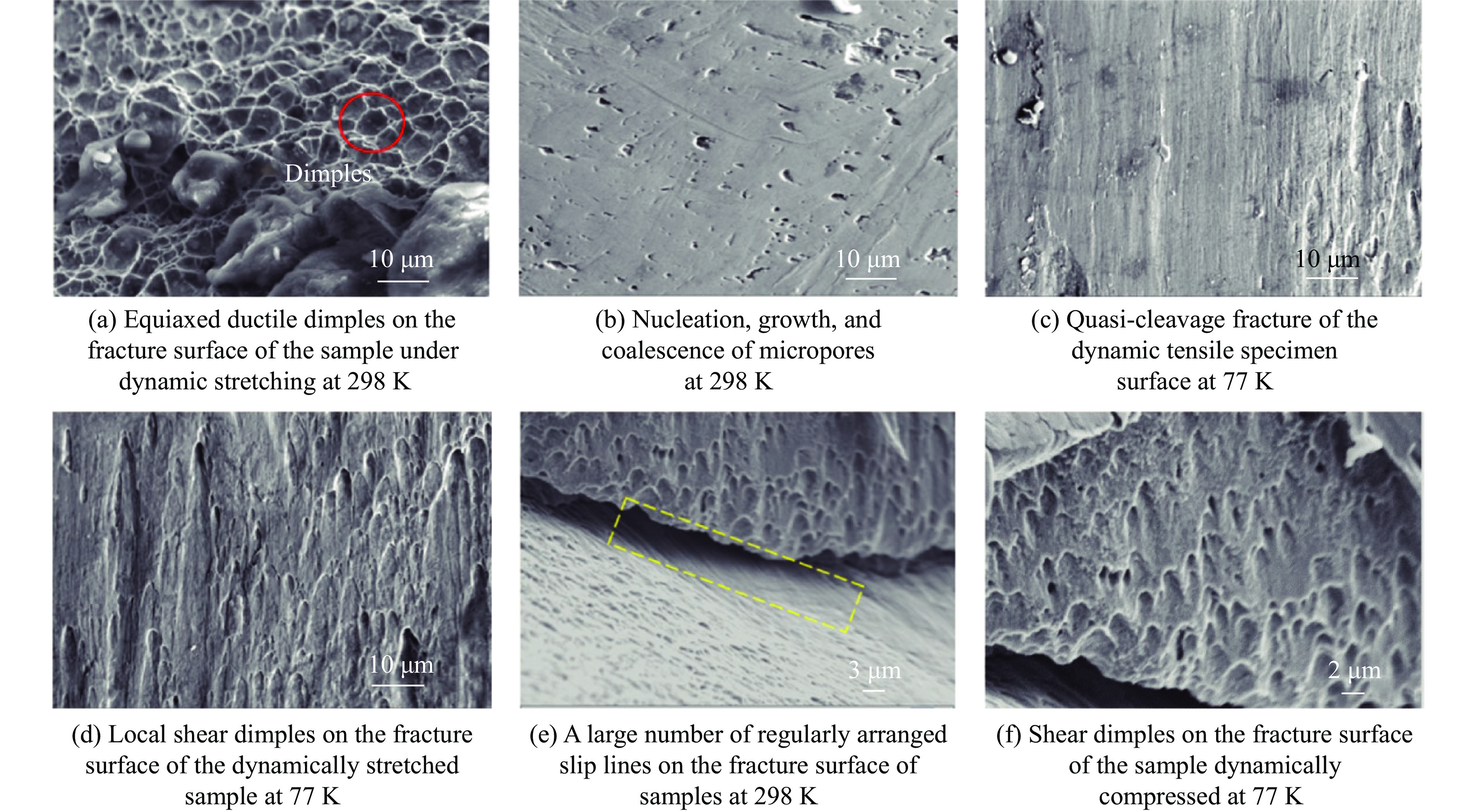
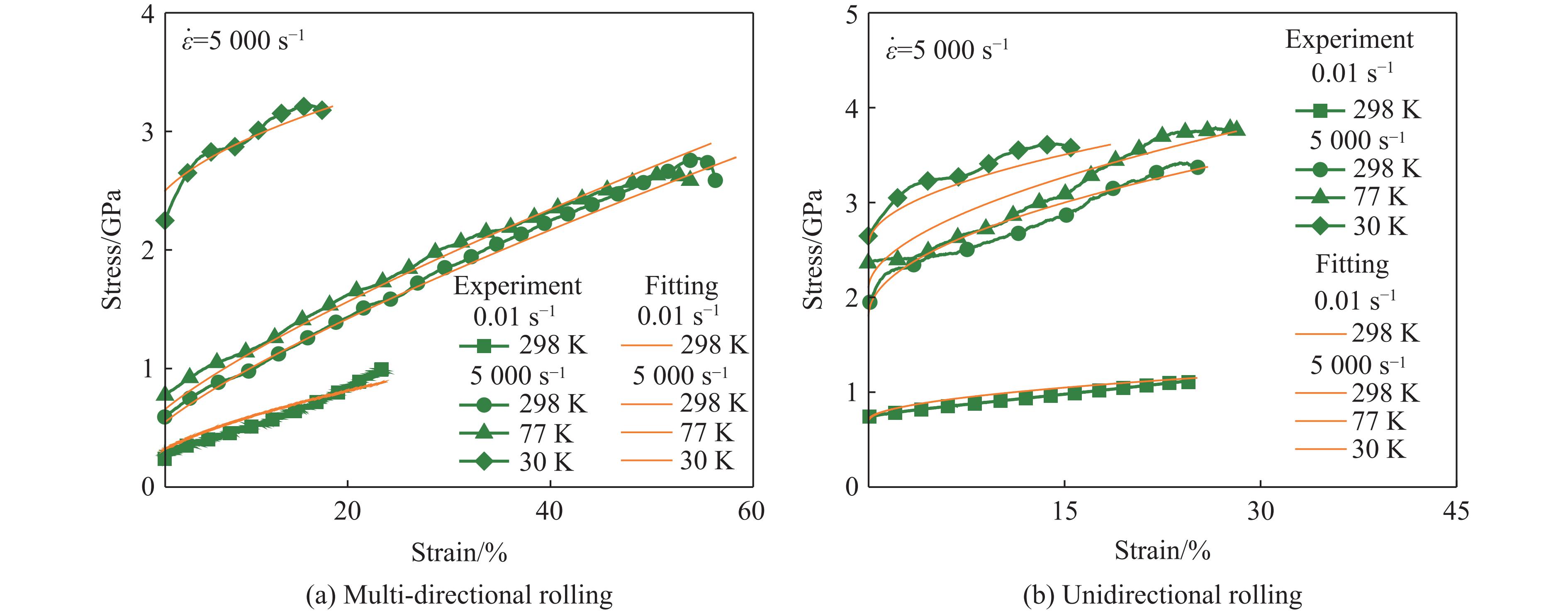
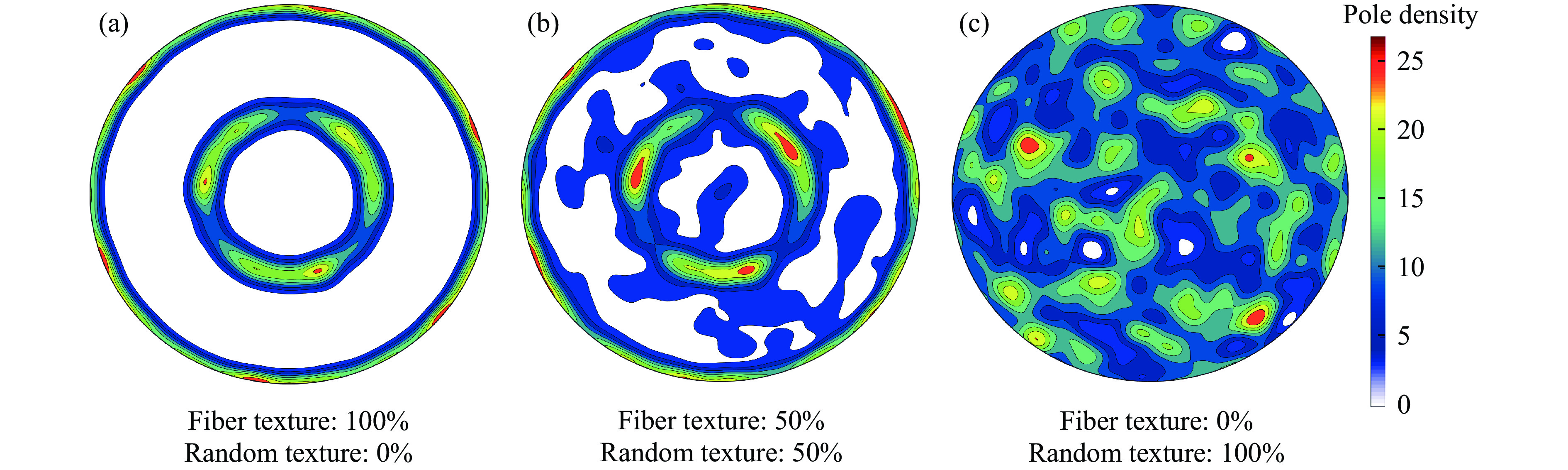
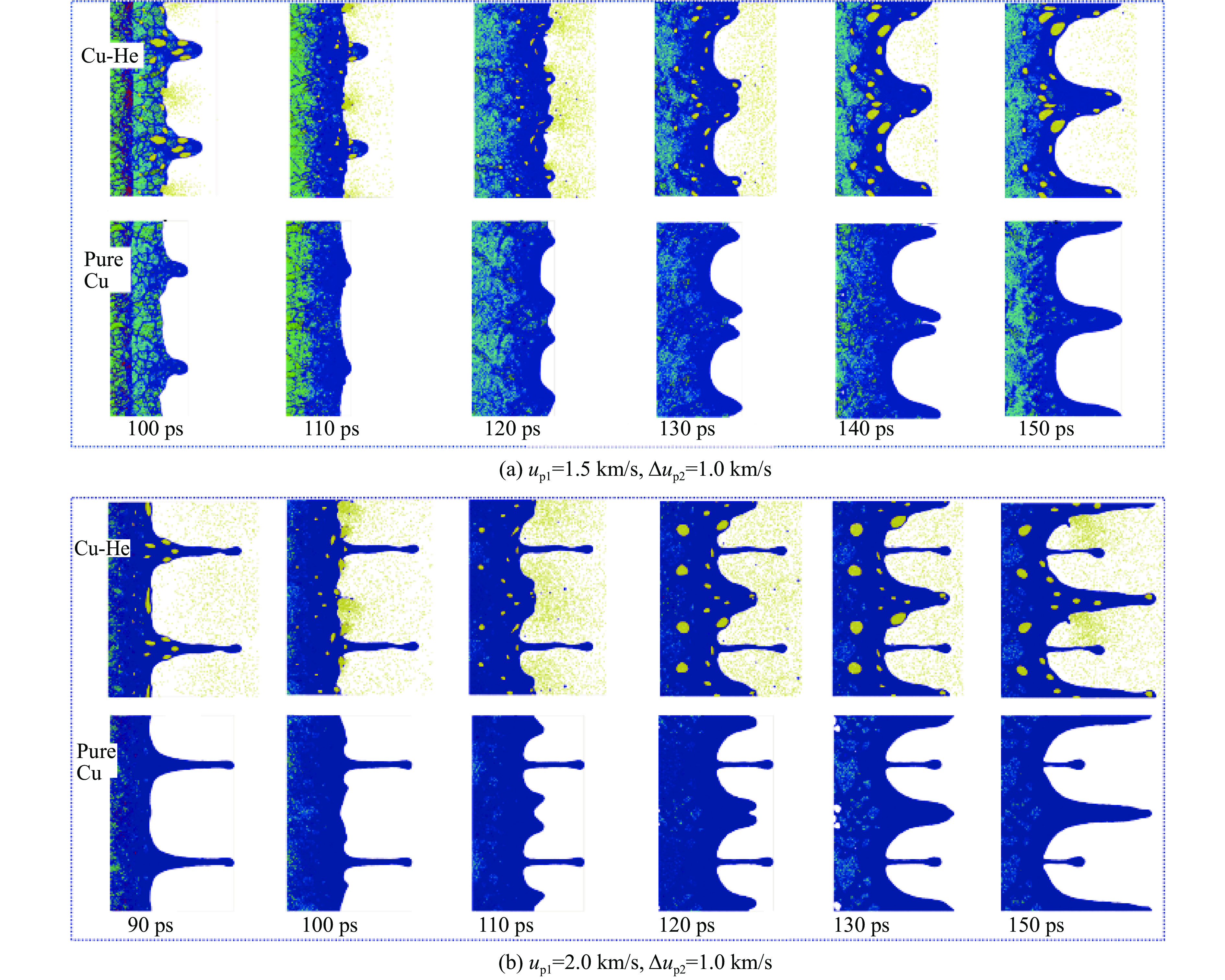
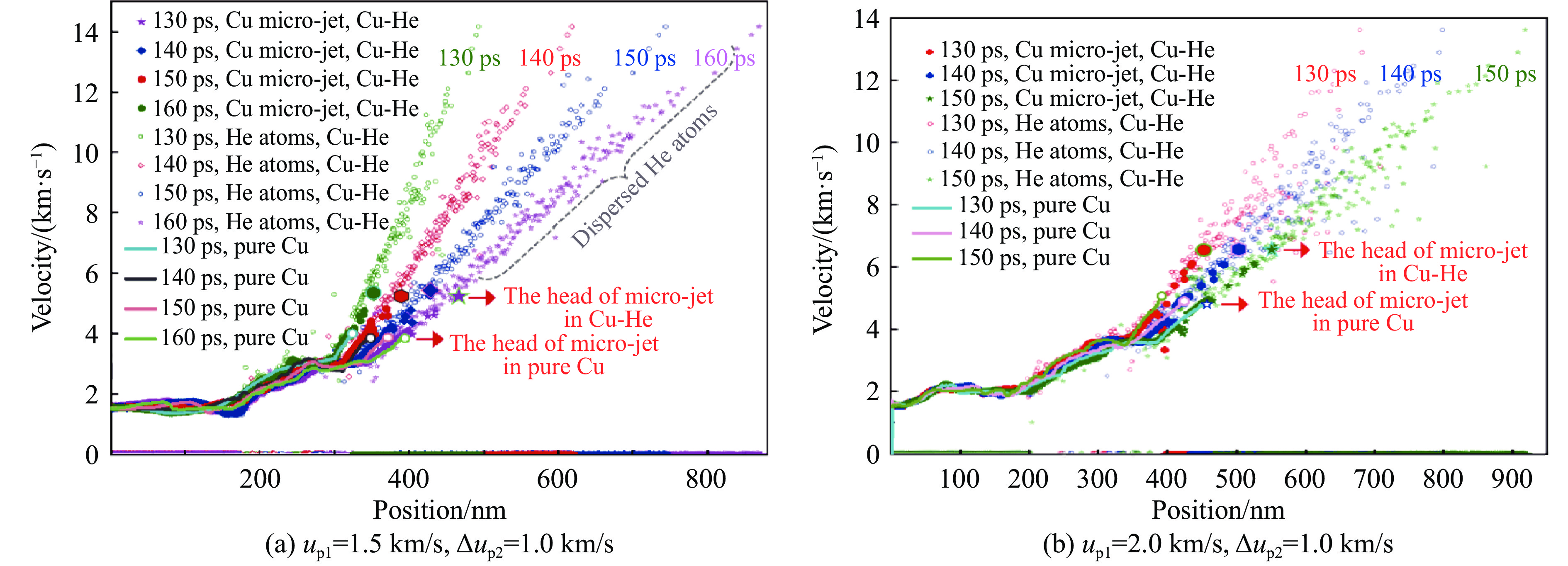
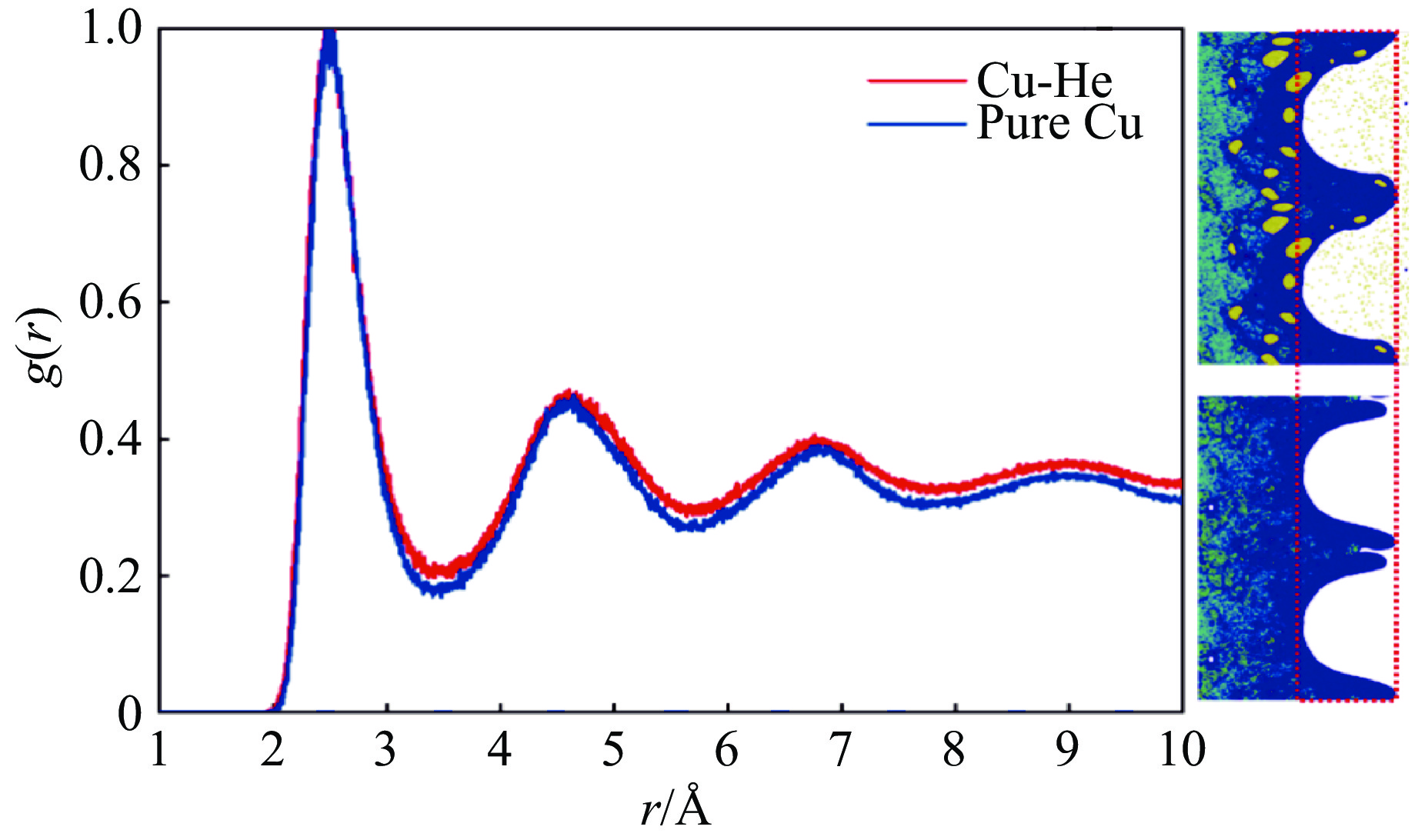
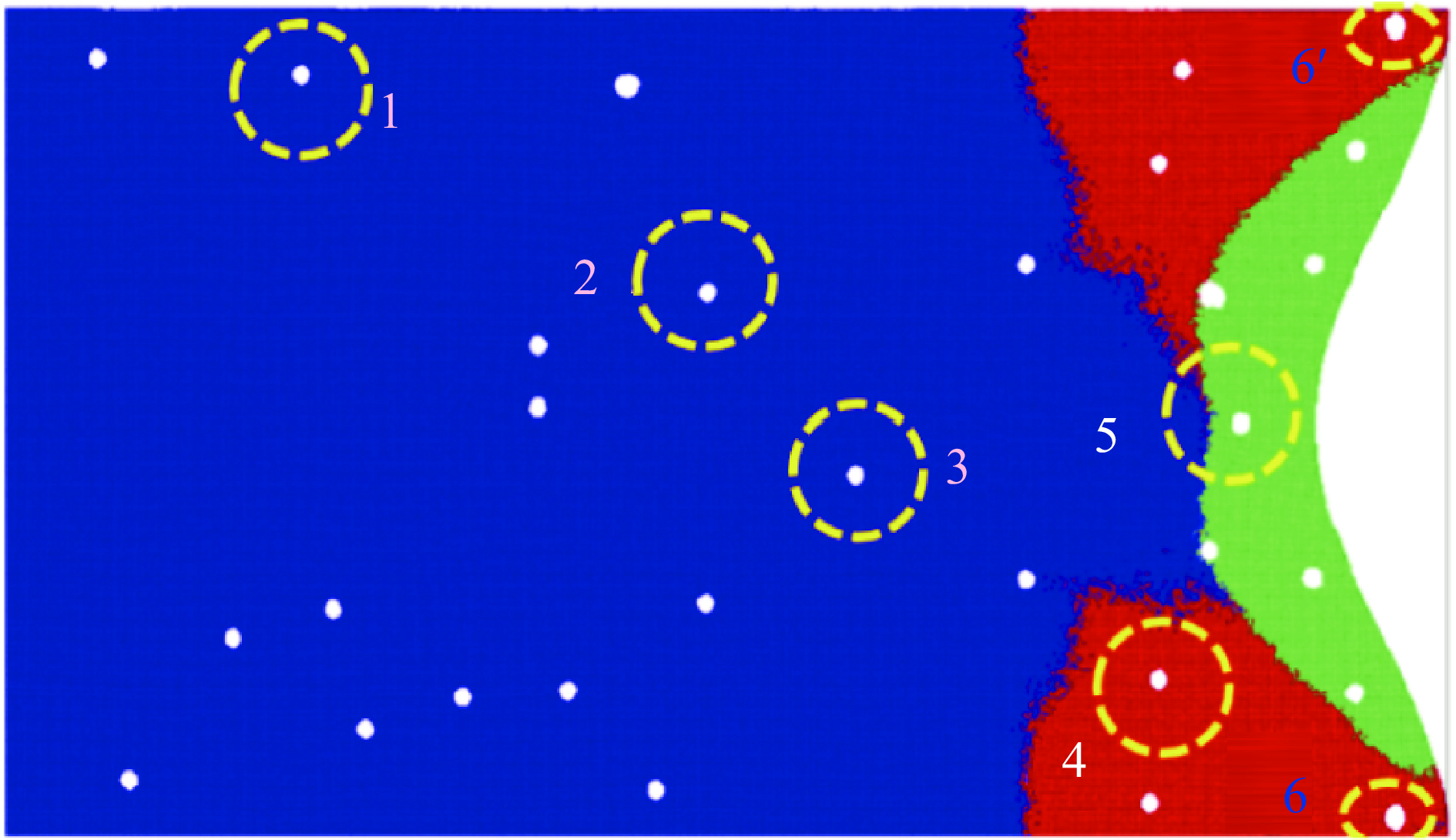
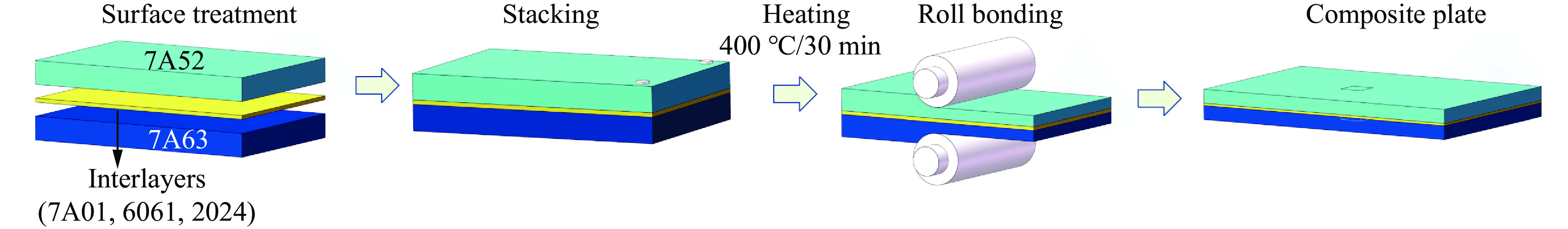
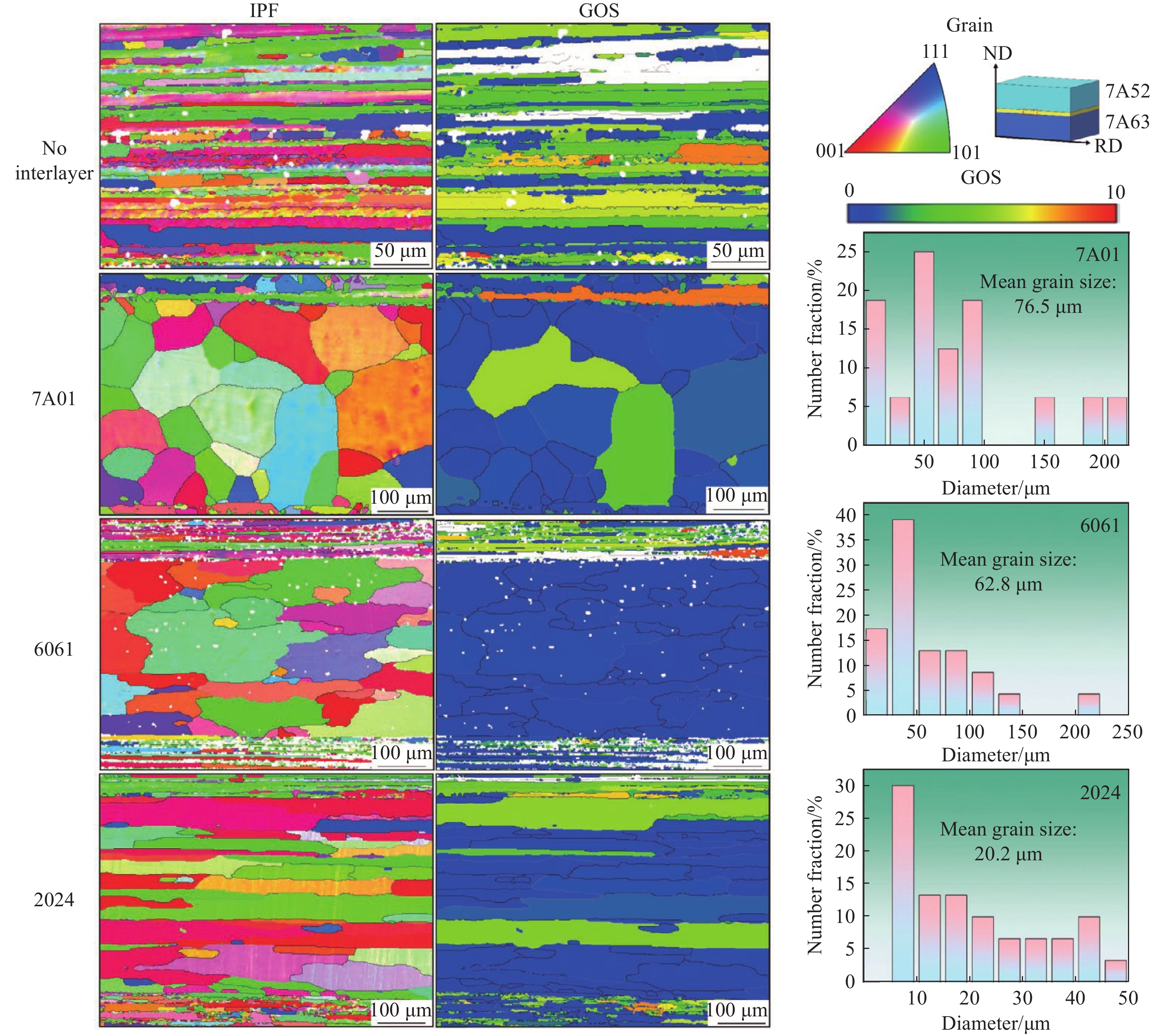
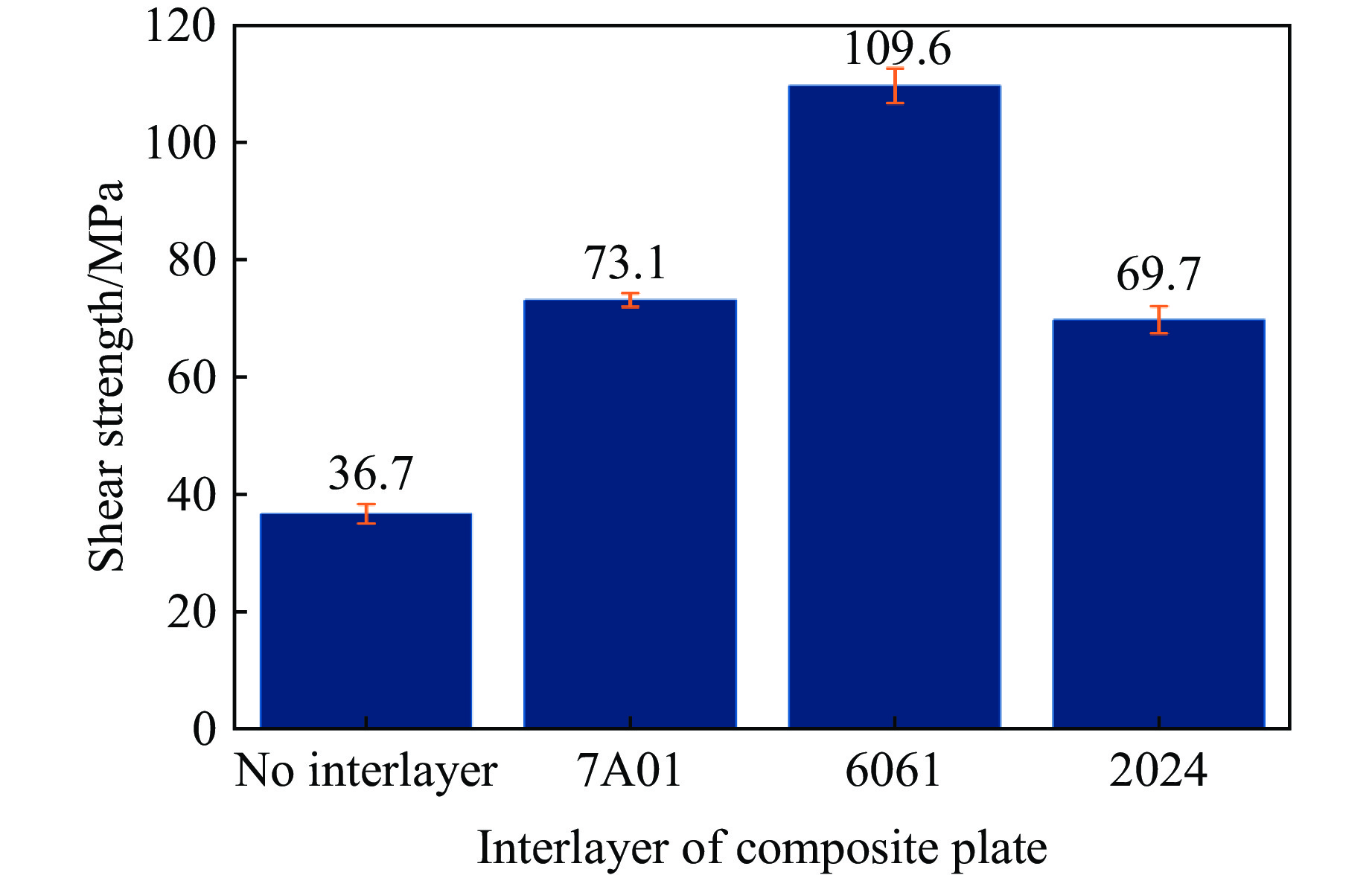
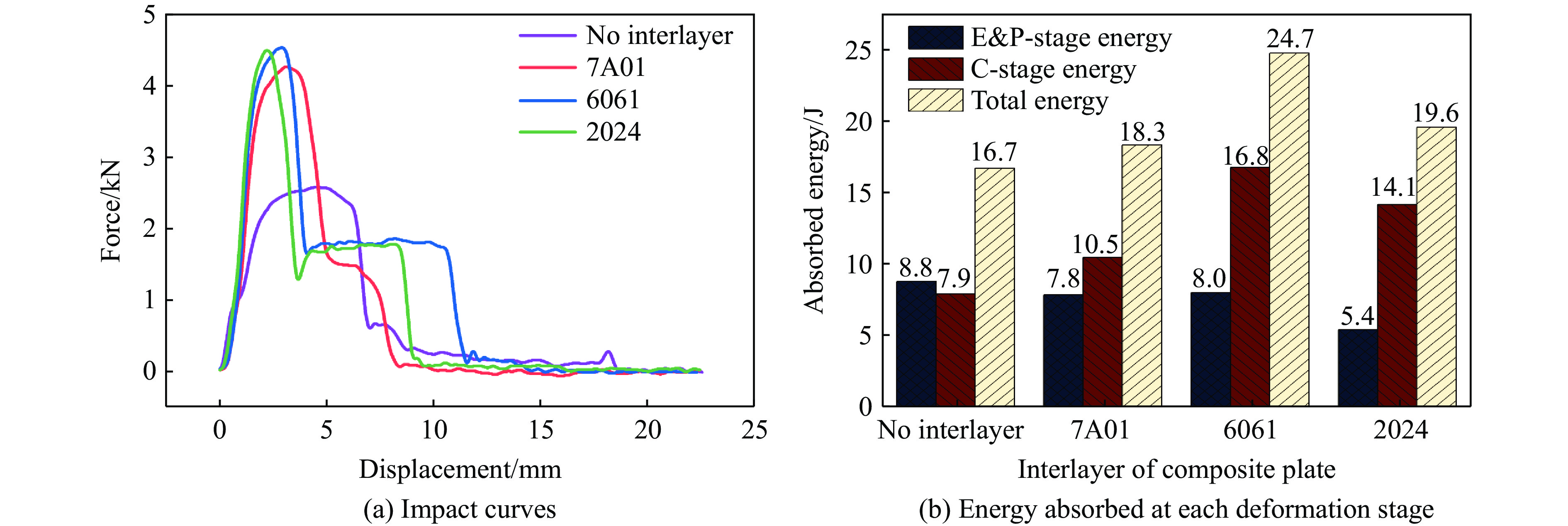
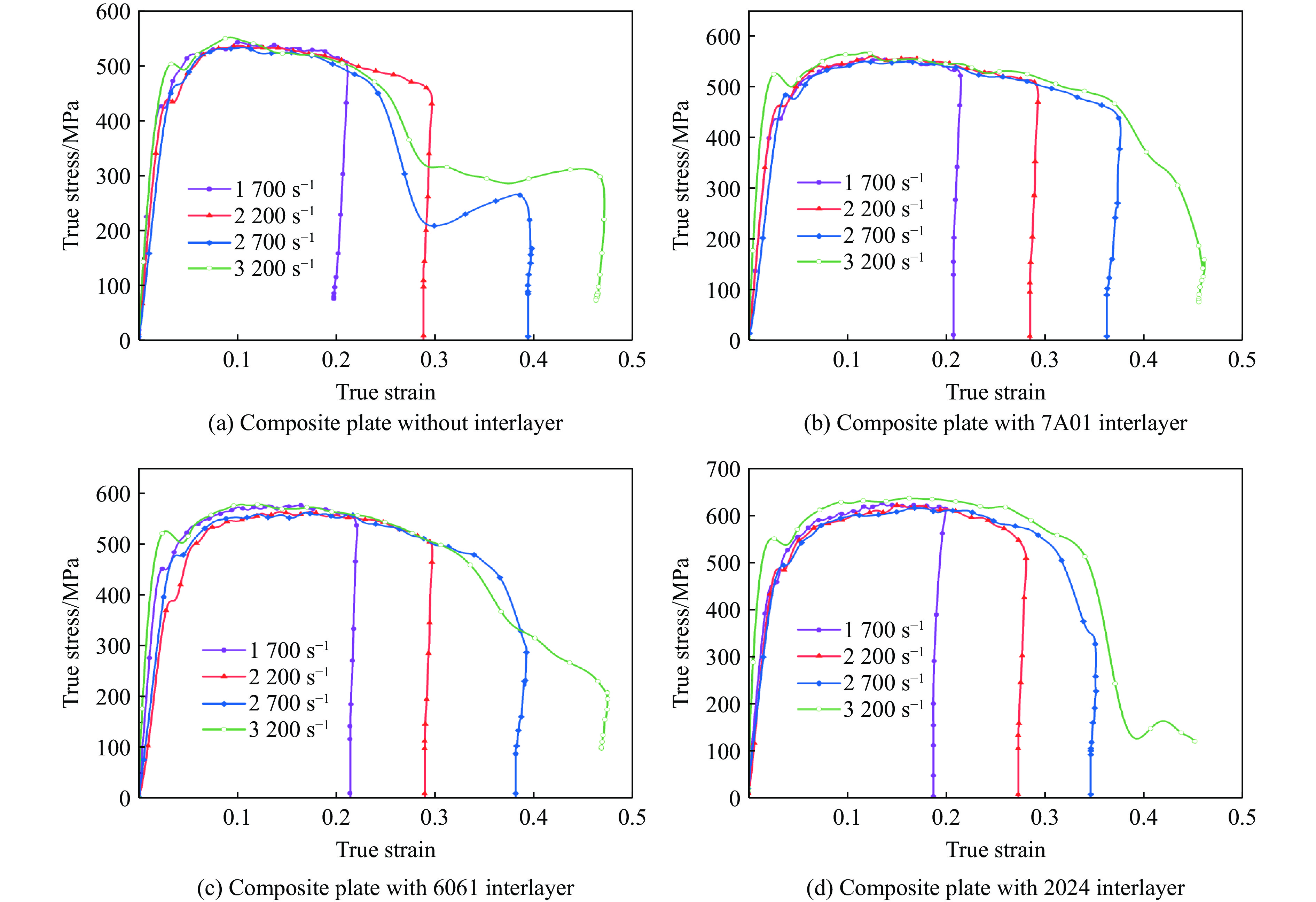
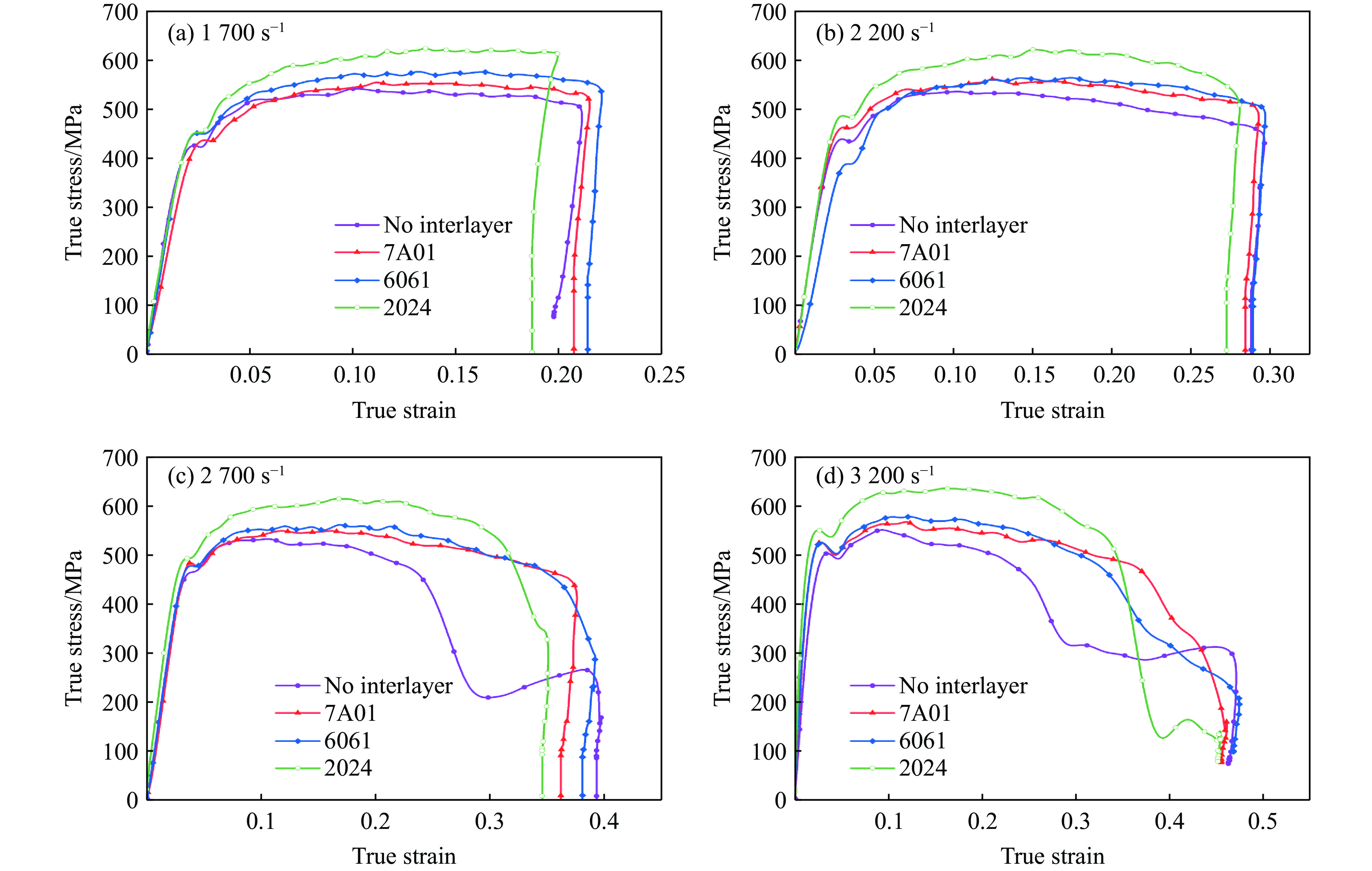
The choice of aluminum alloys used as the interlayers significantly influences the interfacial bonding properties and dynamic impact mechanical properties of 7B53 aluminum alloy composite plates (7A52/interlayer/7A63). In this study, the influence mechanism of different aluminum alloy interlayer materials (7A01, 6061, 2024 aluminum alloy) on the interfacial metallurgical bonding quality and the dynamic mechanical behavior at high strain rates (







Founded in 1987, monthly
Sponsored by:Topical Committee of High Pressure of Chinese Physical Society Sichuan Physical Society
Organized by:Institute of Fluid Physics, CAEP
Editor-in-Chief:MA Yanming
Login in


NewsMore
- 2025 Symposium on Engineering Structure Safety and Protection (First Announcement)
- The 22nd Chinese Conference on High Pressure Science (Third Announcement)
- Notification for the Selection of the Fifth High-Pressure Science Outstanding Young Scholars
- Results of the 2024 Excellent Reviewer Selection for the Journal of High Pressure Physics
- Call for Papers for the Special Issue on Machine Learning and High-Throughput Research of Material Properties under Dynamic Loading
- Notice for the 2024 Shanghai Synchrotron Radiation Large Pressure Machine Experimental Technology Training Course
- Chinese Journal of High Pressure Physics will change from a bimonthly journal to a monthly journal starting in January 2025






