




Progress of Atomistic Simulations for Plastic Bonded Explosives
-
摘要: 回顾了近年来在高聚物黏结炸药(PBX)原子和分子尺度数值模拟方面取得的进展,主要研究领域包括以下6个方面:炸药分子力场、热力学参数计算、耗散/输运性能、相图/相变动力学、动力学响应行为和热点形成机制。针对当前研究现状,介绍了各领域的代表性工作和主要研究成果。目前对PBX炸药的结构和静力学性能已有较充分的认识,但对炸药的动力学响应行为和细观起爆机制尚缺少系统的科学认识,存在一系列挑战性问题,如结构缺陷在爆轰反应后期的形态和表征,以及初始缺陷对爆轰波波形畸变的影响机制。需要将理论计算与实验相结合,以解决爆轰物理领域中的难点问题。Abstract: The recent atomistic simulations of plastic bonded explosive is reviewed in six aspects: the force-field, thermodynamic property, dissipation/transport property, phase transition, constitutive relation and ignition mechanism. In past decades, the structure and mechanical property of PBX are carefully investigated. However, the microscopic defect evolution and hot spot formation mechanisms are unclear. There are a set of challenging problems in detonation physics, such as the defect configuration at the chemical reaction zone, and the detonation wave deformation induced by defect. To investigate them, both atomistic simulation and experiment are required.
-
Key words:
- plastic bonded explosive /
- molecular dynamics /
- thermodynamic properties
-

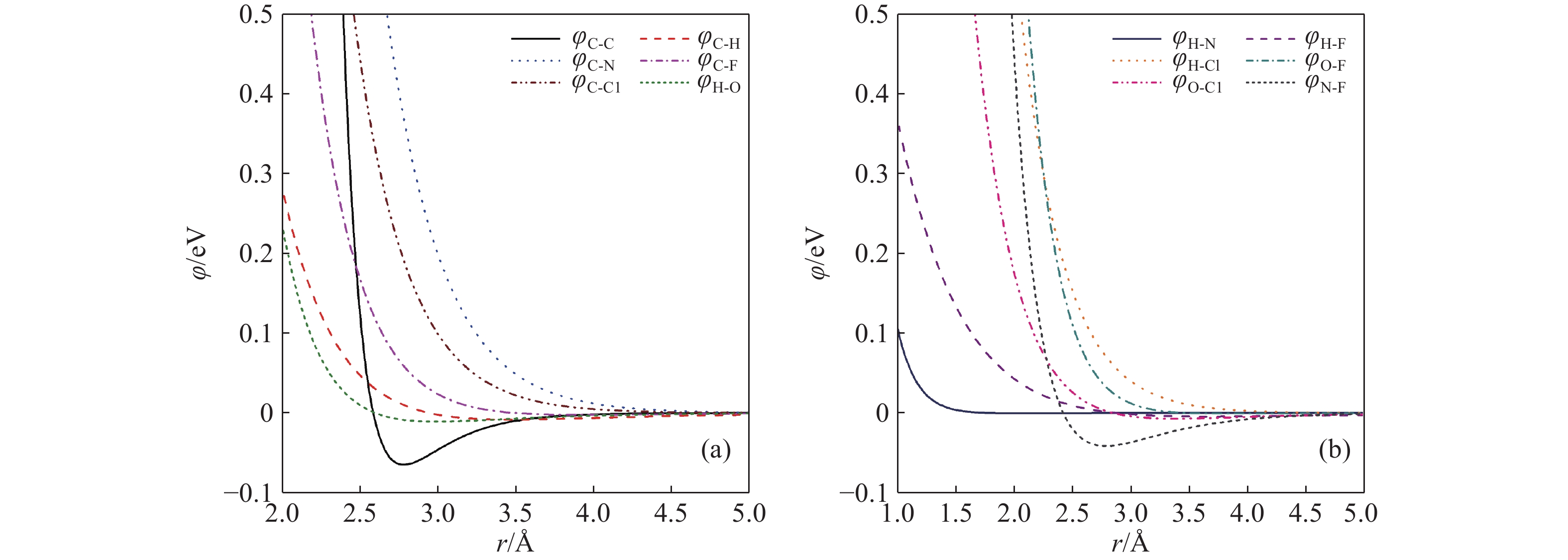
图 1 TATB/氟聚物界面原子势曲线
Figure 1. The potential curves for the TATB/fluoropolymer interface

图 3 (a) RDX多晶,(b) 石蜡包覆RDX,(c) F2311包覆RDX
Figure 3. (a) RDX polycrystal, (b) paraffin coated RDX, (c) F2311 coated RDX

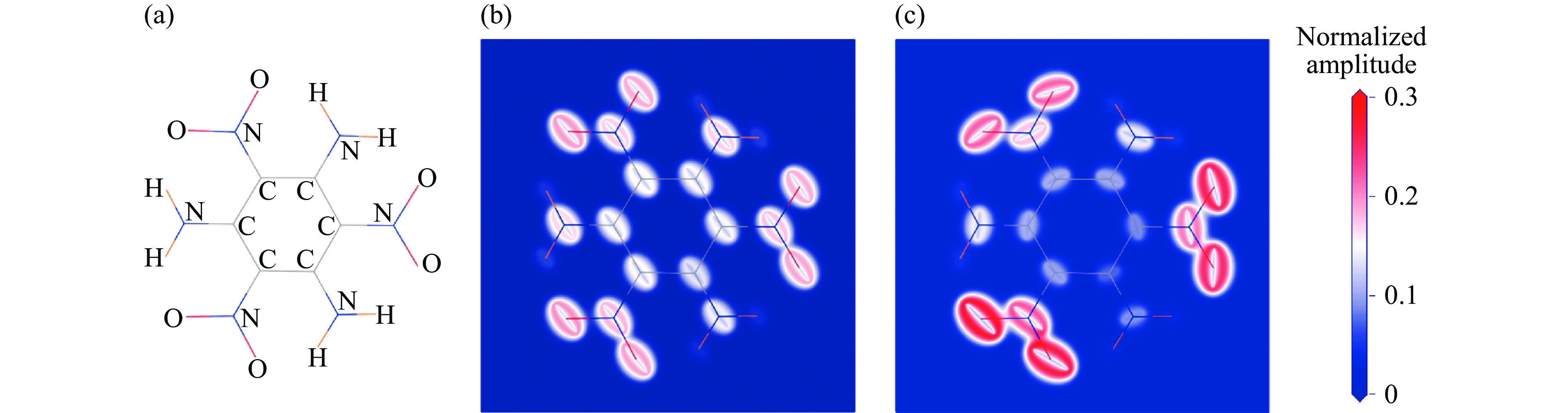
图 4 对TATB热导起关键作用的部分分子振动模式:(a) TATB分子结构,(b) 振动模式1,(c) 振动模式2
Figure 4. Some key vibrational modes for thermal conduction in TATB: (a) TATB molecular structure, (b) the first vibrational mode, (c) the second vibrational mode
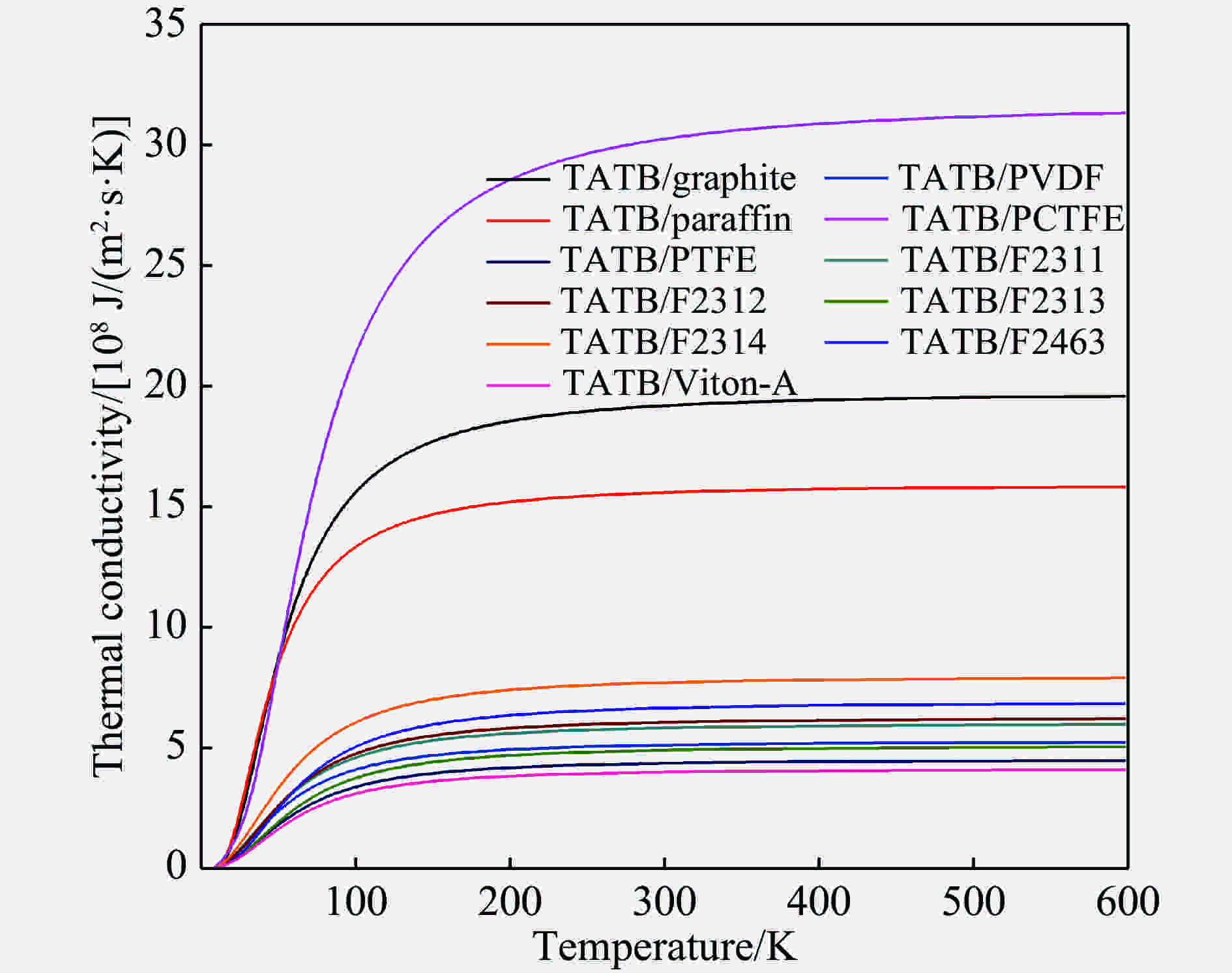
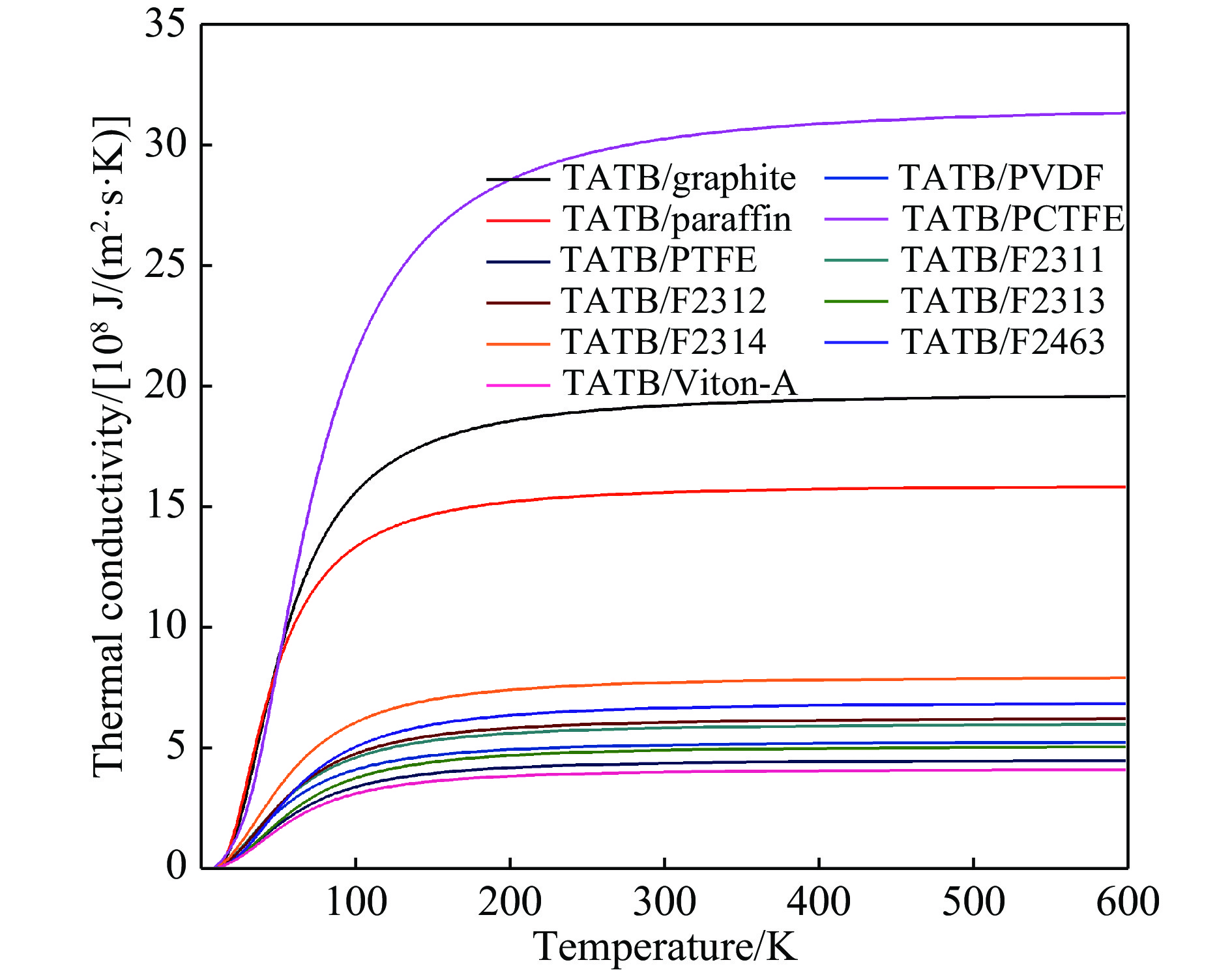
图 5 TATB/添加剂界面热导率的计算结果
Figure 5. The interfacial thermal conductivity vs. temperature for TATB/additive interfaces

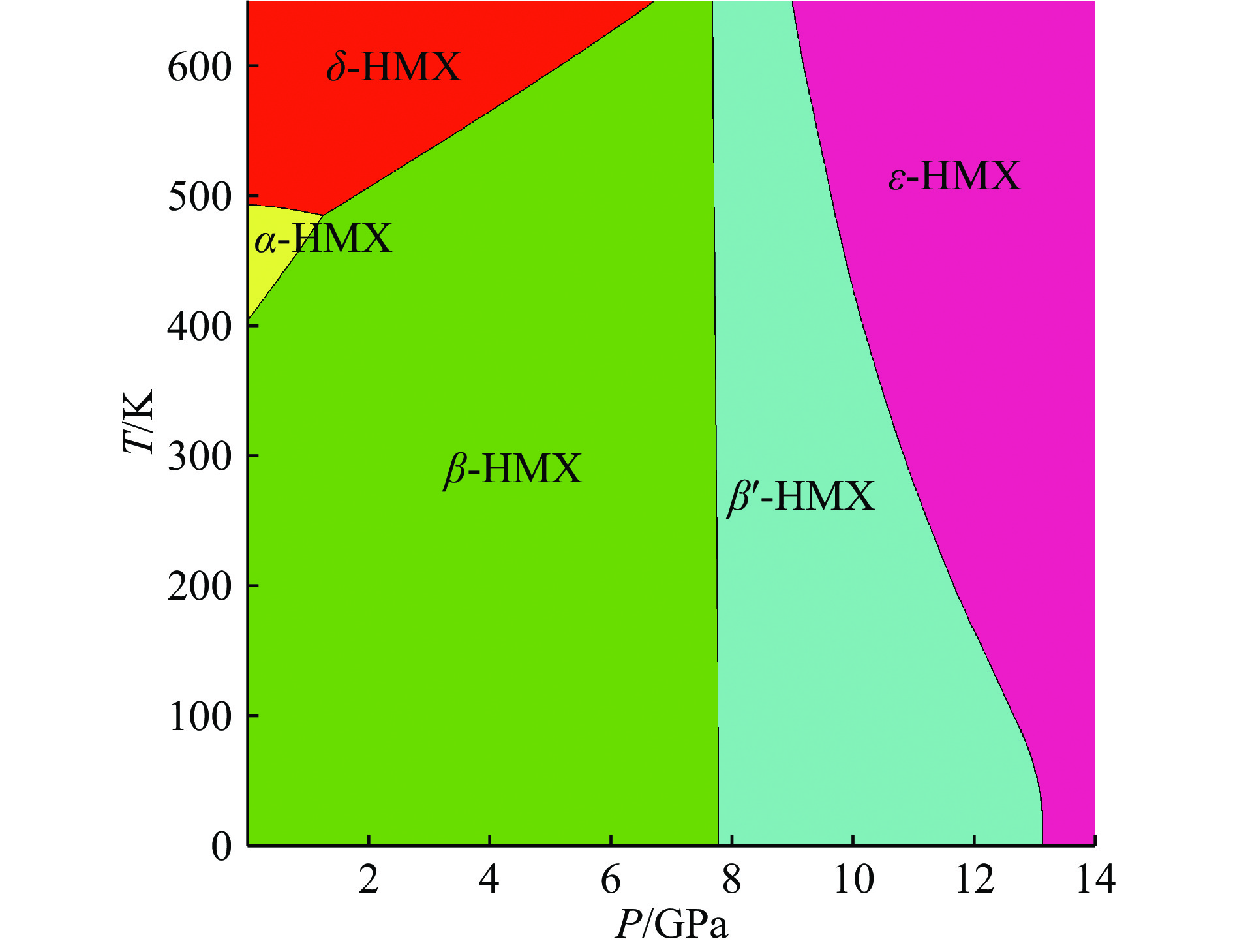
图 6 通过德拜声子理论计算得到的HMX的三相相图(
${\beta '}$ 表示${\beta }$ 相通过声子软化形成的新相)Figure 6. The phase diagram of HMX, obtained by Debye theory (The
${\beta '}$ phase is the phonon soften state of${\beta }$ phase.)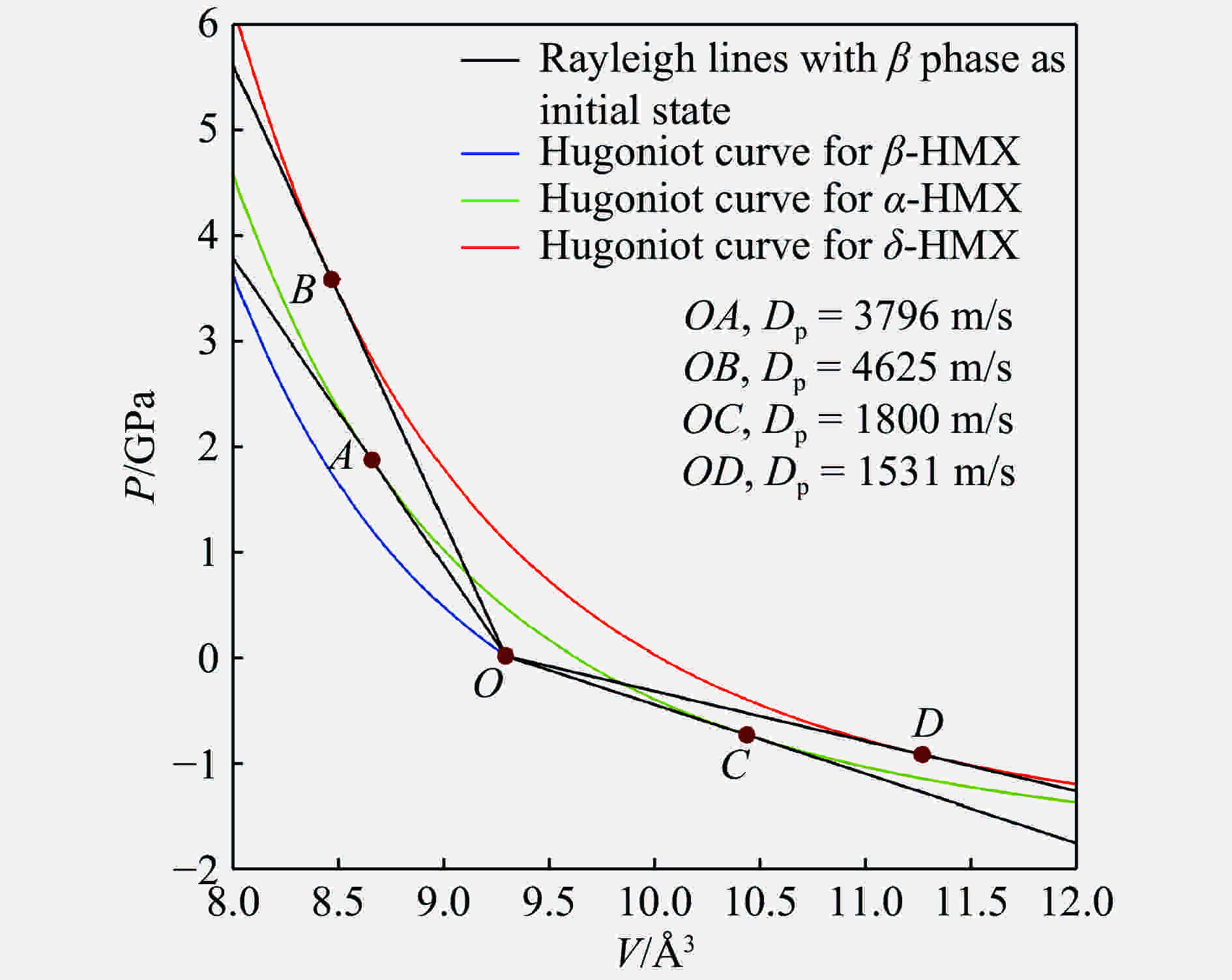
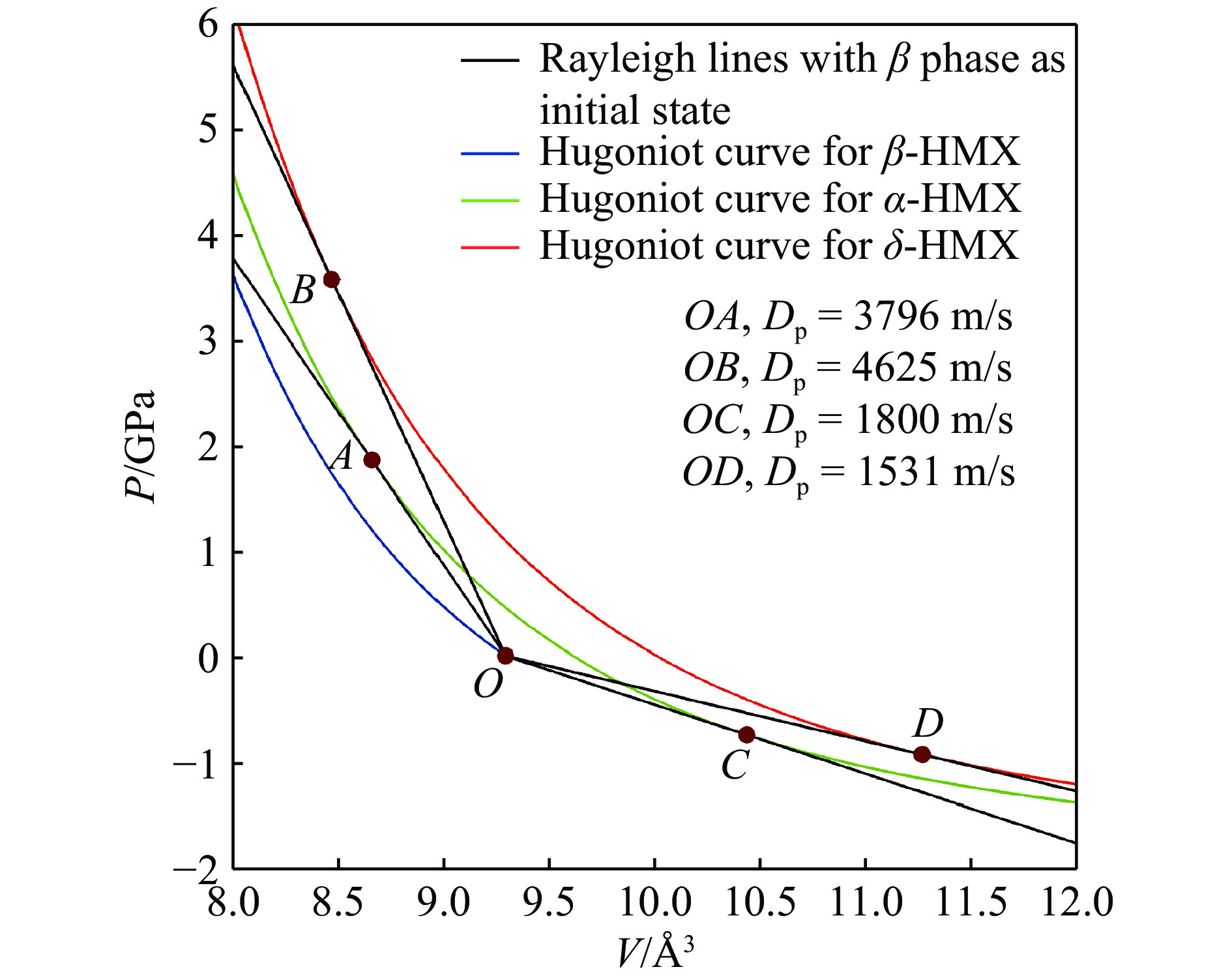
图 7 HMX三相的雨贡纽线和瑞利线
Figure 7. The Hugoniot curves and Rayleigh curve for the three phases of HMX

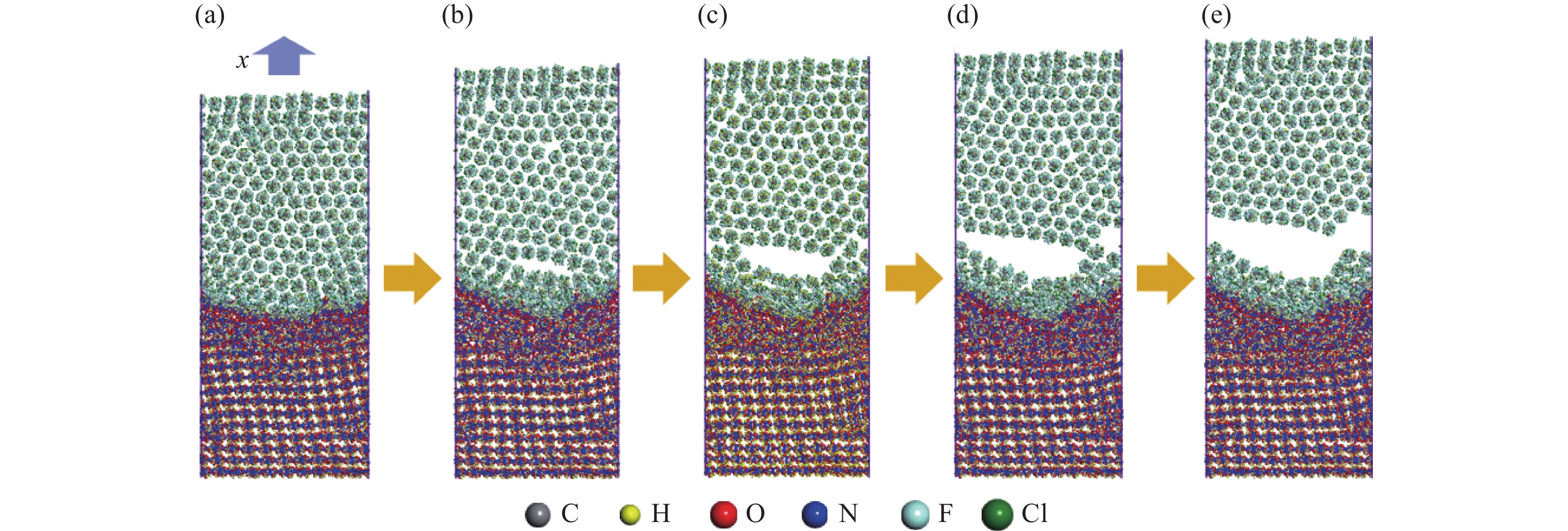
图 8 孔洞塌缩后的场分布:(a) 温度场,(b) 密度场
Figure 8. The temperature field (a) and density field (b) after pore collapsing
-
[1] SORESCU D C, RICE B M, THOMPSON D L. A transferable intermolecular potential for nitramine crystals [J]. The Journal of Physical Chemistry A, 1998, 102(43): 8386–8392. doi: 10.1021/jp9820525 [2] SORESCU D C, RICE B M, THOMPSON D L. Isothermal-isobaric molecular dynamics simulations of 1, 3, 5, 7-tetranitro-1, 3, 5, 7-tetraazacyclooctane (HMX) crystals [J]. The Journal of Physical Chemistry A, 1998, 102(35): 6692–6695. doi: 10.1021/jp981661+ [3] SMITH G D, BHARADWAJ R K. Quantum chemistry based force field for simulations of HMX [J]. The Journal of Physical Chemistry B, 1999, 103(4): 3570–3575. [4] BEDROV D, AYYAGARI C, SMITH G, et al. Molecular dynamics simulations of HMX crystal polymorphs using a flexible molecule force field [J]. Journal of Computer-Aided Materials Design, 2001, 8: 77–85. doi: 10.1023/A:1020046817543 [5] BEDROV D, BORODIN O, SMITH G, et al. A molecular dynamics simulation study of crystalline 1, 3, 5-triamino-2, 4, 6-trinitrobenzene as a function of pressure and temperature [J]. The Journal of Chemical Physics, 2009, 131: 224703. doi: 10.1063/1.3264972 [6] SEWELL T D, MENIKOFF R, BEDROW D, et al. A molecular dynamics simulation study of elastic properties of HMX [J]. The Journal of Chemical Physics, 2003, 119(14): 7417–7426. doi: 10.1063/1.1599273 [7] BEDROV D, SMITH D. Thermal conductivity of molecular fluids from molecular dynamics simulations: application of a new imposed-flux method [J]. The Journal of Chemical Physics, 2000, 113(18): 8080–8084. doi: 10.1063/1.1312309 [8] KROONBLAWD M P, SEWELL T D. Theoretical determination of anisotropic thermal conductivity for crystalline 1, 3, 5-triamino-2, 4, 6- trinitrobenzene (TATB) [J]. The Journal of Chemical Physics, 2013, 139: 074503. doi: 10.1063/1.4816667 [9] KROONBLAWD M P, SEWELL T D. Theoretical determination of anisotropic thermal conductivity for initially defect-free and defective TATB single crystals [J]. The Journal of Chemical Physics, 2014, 141: 184501. doi: 10.1063/1.4901206 [10] GEE R, ROSZAK S, BALASUBRAMANIAN L, et al. Ab initio based force field and molecular dynamics simulations of crystalline TATB [J]. The Journal of Chemical Physics, 2004, 120(15): 7059–7066. doi: 10.1063/1.1676120 [11] SONG H J, ZHANG Y G, LI H, et al. All-atom, non-empirical, and tailor-made force field for α-RDX from first principles [J]. RSC Advances, 2014, 4(76): 40518–40533. doi: 10.1039/C4RA07195F [12] SUN H. COMPASS: An ab initio force-field optimized for condensed-phase applications-overview with details on alkane and benzene compounds [J]. The Journal of Physical Chemistry B, 1998, 102: 7338–7364. doi: 10.1021/jp980939v [13] BUNTE S, SUN H. Molecular modeling of energetic materials: the parameterization and validation of nitrate esters in the COMPASS force field [J]. The Journal of Physical Chemistry B, 2000, 104(11): 2477–2489. doi: 10.1021/jp991786u [14] TERSOFF J. Empirical interatomic potential for carbon, with applications to amorphous carbon [J]. Physical Review Letters, 1988, 61(25): 2879–2882. doi: 10.1103/PhysRevLett.61.2879 [15] LONG Y, LIU Y G, NIE F D, et al. Force-field derivation and atomistic simulation of HMX/graphite interface and polycrystal systems [J]. Communications in Theoretical Physics, 2012, 57(1): 102–114. doi: 10.1088/0253-6102/57/1/16 [16] LONG Y, LIU Y G, NIE F D, et al. The force-field derivation and atomistic simulation of HMX-fluoropolymer mixture explosives [J]. Colloid & Polymer Science, 2012, 290(18): 1855–1866. [17] LONG Y, LIU Y G, NIE F D, et al. Force-field derivation and atomistic simulation of HMX-TATB-graphite mixture explosives [J]. Modelling and Simulation in Materials Science and Engineering, 2012, 20(6): 065010. doi: 10.1088/0965-0393/20/6/065010 [18] LONG Y, CHEN J. The force-field derivation and application of explosive/additive interfaces [J]. Modelling and Simulation in Materials Science and Engineering, 2016, 24(7): 075013. doi: 10.1088/0965-0393/24/7/075013 [19] LONG Y, CHEN J. Theoretical study of the interfacial force-field, thermodynamic property, and heat stress for plastic bonded explosives [J]. The Journal of Physical Chemistry C, 2017, 121(5): 2778–2788. doi: 10.1021/acs.jpcc.6b11203 [20] STRACHAN A, DUIN A, GODDARD W, et al. Shock waves in high-energy materials: the initial chemical events in nitramine RDX [J]. Physical Review Letters, 2003, 91: 098301. doi: 10.1103/PhysRevLett.91.098301 [21] LIU L, LIU Y, GODDARD W, et al. ReaxFF-lg: correction of the ReaxFF reactive force field for London dispersion, with applications to the equations of state for energetic materials [J]. The Journal of Physical Chemistry A, 2011, 115(40): 11016–11022. doi: 10.1021/jp201599t [22] ZHANG L, ZYBIN S V, DUIN A, et al. Carbon cluster formation during thermal decomposition of octahydro-1, 3, 5, 7-tetranitro-1, 3, 5, 7- tetrazocine and 1, 3, 5-triamino-2, 4, 6-trinitrobenzene high explosives from ReaxFF reactive molecular dynamics simulations [J]. The Journal of Physical Chemistry A, 2009, 113(40): 10619–10640. doi: 10.1021/jp901353a [23] AN Q, ZYBIN S V, GORRDAR W A, et al. Elucidation of the dynamics for hot-spot initiation at nonuniform interfaces of highly shocked materials [J]. Physical Review B: Condensed Matter, 2011, 84: 220101. doi: 10.1103/PhysRevB.84.220101 [24] AN Q, GORRDAR W A, ZYBIN S V, et al. Highly shocked polymer bonded explosives at a nonplanar interface: hot-spot formation leading to detonation [J]. The Journal of Physical Chemistry C, 2013, 117(50): 26551–26561. doi: 10.1021/jp404753v [25] CHERUKARA M J, WOOD M A, KOBER E M, et al. Ultra-fast chemistry under non-equilibrium conditions and the shock to deflagration transition at the nanoscale [J]. The Journal of Physical Chemistry C, 2015, 119: 22008–22015. doi: 10.1021/acs.jpcc.5b05362 [26] 肖鹤鸣, 居学海. 高能体系中的分子间相互作用 [M]. 北京: 科学出版社, 2004. [27] 肖鹤鸣, 许晓娟, 邱玲. 高能量密度材料的理论设计 [M]. 北京: 科学出版社, 2008. [28] 肖继军, 朱卫华, 朱伟, 等. 高能材料分子动力学 [M]. 北京: 科学出版社, 2013. [29] XIAO J J, WANG W R, CHEN J, et al. Study on the relations of sensitivity with energy properties for HMX and HMX-based PBXs by molecular dynamics simulation [J]. Physica B: Condensed Matter, 2012, 407(17): 3504–3509. doi: 10.1016/j.physb.2012.05.010 [30] CAO Q, XIAO J J, GAO P, et al. Molecular dynamics simulations for CL-20/TNT co-crystal based polymer-bonded explosives [J]. Journal of Theoretical and Computational Chemistry, 2017, 16: 1750072. doi: 10.1142/S0219633617500729 [31] ZERILLI F J, KUKLJA M. Ab initio equation of state of the organic molecular crystal: β-Octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine [J]. The Journal of Physical Chemistry A, 2010, 114(16): 5372–5376. doi: 10.1021/jp911767q [32] VALENZANO L, SLOUGH W J, PERGER W. Accurate prediction of second-order elastic constants from first principles: PETN and TATB [J]. AIP Conference Proceedings, 2012, 1426: 1191–1194. [33] CUI H, JI G F, CHEN X, et al. Phase transitions and mechanical properties of octahydro-1, 3, 5, 7-tetranitro-1, 3, 5, 7- tetrazocine in different crystal phases by molecular dynamics simulation [J]. Journal of Chemical & Engineering Data, 2010, 55(9): 3121–3129. [34] LONG Y, LIU Y G, NIE F D, et al. Theoretical study of breaking and slipping processes for HMX/graphite interface [J]. Applied Surface Science, 2012, 258(7): 2384–2392. doi: 10.1016/j.apsusc.2011.10.052 [35] QIU L, XIAO H M. Molecular dynamics study of binding energies, mechanical properties, and detonation performances of bicyclo-HMX-based PBXs [J]. Journal of Hazardous Material, 2009, 164(1): 329–336. doi: 10.1016/j.jhazmat.2008.08.030 [36] XIAO J, HUANG H, LI J S, et al. A molecular dynamics study of interface interactions and mechanical properties of HMX-based PBXs with PEG and HTPB [J]. Journal of Molecular Structure: THEOCHEM, 2008, 851(1): 242–248. [37] XIAO J, ZHANG H, HUANG H, et al. NPT ensemble MD simulation investigation on the mechanical properties of HMX/F2311 polymer-bonded explosive [J]. Chinese Journal of Chemistry, 2008, 26(11): 1969–1972. doi: 10.1002/cjoc.v26:11 [38] JAIDANN M, LUSSIER L S, BOUAMOUL A, et al. Effects of interface interactions on mechanical properties in RDX-based PBXs HTPB-DOA: molecular dynamics simulations [C]//Computational Science-ICCS 2009. Heidelberg: Springer-Berlin, 2009: 131-140. [39] JAIDANN M, LUSSIER L S, BOUAMOUL A, et al. Atomistic studies of RDX and FOX-7-based plastic-bonded explosives: molecular dynamics simulation [J]. Procedia Computer Science, 2011, 4: 1177–1185. doi: 10.1016/j.procs.2011.04.126 [40] KUBO R, TODA M, HASHITSUME N. Statistical physics II: nonequilibrium statistical mechanics [M]. Springer-Verlag, 1997. [41] PARLINSKI K, LI Z Q, KAWAZOE Y. First-principles determination of the soft mode in cubic ZrO2 [J]. Physical Review Letters, 1997, 78(21): 4063–4066. doi: 10.1103/PhysRevLett.78.4063 [42] MARADUDIN A, FEIN A. Scattering of neutrons by an anharmonic crystal [J]. Physical Review, 1962, 128(6): 2589–2608. doi: 10.1103/PhysRev.128.2589 [43] TOGO A, CHAPUT L, TANAKA I. Distributions of phonon lifetimes in Brillouin zones [J]. Physical Review B: Condensed Matter, 2015, 91: 094306. doi: 10.1103/PhysRevB.91.094306 [44] MÜLLER-PLATHE F. Reversing the perturbation in nonequilibrium molecular dynamics: an easy way to calculate the shear viscosity of fluids [J]. Physical Review E, 1999, 59: 4894–4898. [45] BEDROV D, SMITH G D, SEWELL T D. Thermal conductivity of liquid octahydro-1, 3, 5, 7-tetranitro-1, 3, 5, 7-tetrazocine (HMX) from molecular dynamics simulations [J]. Chemical Physics Letters, 2000, 324: 64–68. doi: 10.1016/S0009-2614(00)00559-5 [46] KROONBLAWD M P, SEWELL T D. Predicted anisotropic thermal conductivity for crystalline 1, 3, 5-triamino-2, 4, 6-trinitobenzene (TATB): temperature and pressure dependence and sensitivity to intramolecular force field terms [J]. Propellants, Explosives, Pyrotechnics, 2016, 41(3): 502–513. doi: 10.1002/prep.v41.3 [47] LONG Y, CHEN J, LIU Y G, et al. A direct method to calculate thermal conductivity and its application in solid HMX [J]. Journal of Physics: Condensed Matter, 2010, 22: 185404. doi: 10.1088/0953-8984/22/18/185404 [48] LONG Y, LIU Y G, NIE F D, et al. A method to calculate the thermal conductivity of HMX under high pressure [J]. Philosophical Magazine, 2012, 92(8): 1023–1045. doi: 10.1080/14786435.2011.637981 [49] LONG Y, CHEN J. Theoretical study of the phonon-phonon scattering mechanism and the thermal conductive coefficients for energetic material [J]. Philosophical Magazine, 2017, 97: 2575–2595. doi: 10.1080/14786435.2017.1343962 [50] LONG Y, CHEN J. A theoretical study of wave dispersion and thermal conduction for HMX/additive interfaces [J]. Modelling and Simulation in Materials Science and Engineering, 2014, 22: 035013. doi: 10.1088/0965-0393/22/3/035013 [51] LONG Y, CHEN J. Theoretical study of the phonon spectrum, phonon refraction and thermodynamic properties for explosive/additive interfaces [J]. Modelling and Simulation in Materials Science and Engineering, 2018, 26: 015002. doi: 10.1088/1361-651X/aa944d [52] BEDROV D, SMITH G D, SEWELL T D. Temperature-dependent shear viscosity coefficient of octahydro-1, 3, 5, 7-tetranitro-1, 3, 5, 7-tetrazocine (HMX): a molecular dynamics simulation study [J]. The Journal of Chemical Physics, 2000, 112(16): 7203–7208. doi: 10.1063/1.481285 [53] LONG Y, CHEN J. The heat dissipation model and desensitizing mechanism of the HMX/additive interfaces: a theoretical investigation based on linear response theory [J]. Modelling and Simulation in Materials Science and Engineering, 2013, 21: 055025. doi: 10.1088/0965-0393/21/5/055025 [54] KHOLOD Y, OKOVYTYY S, KURAMSHINA G, et al. An analysis of stable forms of CL-20: a DFT study of conformational transitions, infrared and Raman spectra [J]. Journal of Molecular Structure, 2007, 843: 14–25. doi: 10.1016/j.molstruc.2006.12.031 [55] XU X J, ZHU W H, XIAO H M. DFT Studies on the four polymorphs of crystalline CL-20 and the influences of hydrostatic pressure on ε-CL-20 crystal [J]. The Journal of Physical Chemistry B, 2007, 111(8): 2090–2097. doi: 10.1021/jp066833e [56] BRAND H V, RABIE R L, FUNK D J, et al. Theoretical and experimental study of the vibrational spectra of the α, β and δ phases of octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (HMX) [J]. The Journal of Physical Chemistry B, 2002, 106(41): 10594–10604. doi: 10.1021/jp020909z [57] MUNDAY L B, CHUNG P W, RICE B M, et al. Simulations of high-pressure phases in RDX [J]. The Journal of Physical Chemistry B, 2011, 115(15): 4378–4386. doi: 10.1021/jp112042a [58] LU L Y, WEI D Q, CHEN X R. The pressure-induced phase transition of the solid β-HMX [J]. Molecular Physics, 2009, 107(22): 2373–2385. doi: 10.1080/00268970903313642 [59] SMILOWITZ L, HENSON B F, ASAY B W, et al. The β-δ phase transition in the energetic nitramine-octahydro-1, 3, 5, 7-tetranitro-1, 3, 5, 7-tetrazocine: kinetics [J]. The Journal of Chemical Physics, 2002, 117(8): 3789–3798. doi: 10.1063/1.1495399 [60] SMILOWITZ L, HENSON B F, ASAY B W, et al. The β-δ phase transition in the energetic nitramine-octahydro-1, 3, 5, 7-tetranitro-1, 3, 5, 7-tetrazocine: thermodynamics [J]. The Journal of Chemical Physics, 2002, 117(8): 3780–3788. doi: 10.1063/1.1495398 [61] LEVITAS V I, HENSON B F, SMILOWITZ L B, et al. Solid-solid phase transformation via internal stress-induced virtual melting, significantly below the melting temperature. application to HMX energetic crystal [J]. The Journal of Physical Chemistry B, 2006, 110(20): 10105–10119. doi: 10.1021/jp057438b [62] LI J, BRILL T B. Kinetics of solid polymorphic phase transitions of CL-20 [J]. Propellants, Explosives, Pyrotechnics, 2007, 32(4): 326–330. doi: 10.1002/(ISSN)1521-4087 [63] MATHEW N, KROONBLAWD M, SEWELL T D, et al. Predicted melt curve and liquid-state transport properties of TATB from molecular dynamics simulations [J]. Molecular Simulation, 2018, 44: 613–622. doi: 10.1080/08927022.2017.1418084 [64] LONG Y, CHEN J. Theoretical study of phonon density of states, thermodynamic properties and phase transitions for HMX [J]. Philosophical Magazine, 2014, 94: 2656–2677. doi: 10.1080/14786435.2014.927598 [65] LONG Y, CHEN J. Theoretical study of the thermodynamic properties, phase transition wave, and phase transition velocity for octahydro-1, 3, 5, 7- tetranitro-1, 3, 5, 7-tetrazocine [J]. Journal of Applied Physics, 2015, 118: 115901. doi: 10.1063/1.4930812 [66] LONG Y, CHEN J. A theoretical study of the stress relaxation in HMX on the picosecond time scale [J]. Modelling and Simulation in Materials Science and Engineering, 2015, 23: 085001. doi: 10.1088/0965-0393/23/8/085001 [67] KURY J W, HORNIG H C, LEE E L, et al. Metal accelaration by chemical explosives [C]//Proceedings of the 4th International Symposium on Detonation. Maryland: White Oak, 1966: 3–13. [68] WANG L L, ZHU X X, SHI S Q. An impact dynamics investigation on some problems in bird strike on windshield of high speed aircrafts [J]. Chinese Journal of Aeronautics, 1991, 12(3): 27–33. [69] 周风华, 王礼立, 胡时胜. 有机玻璃在高应变率下的损伤型非线性粘弹性本构关系及破坏准则 [J]. 爆炸与冲击, 1992, 12(4): 333–341.ZHOU F H, WANG L L, HU S S. A damage-modified nonlinear visco-elastic constitutive relation and its failure criterion of PMMA at high strain rates [J]. Explosion and Shock Waves, 1992, 12(4): 333–341. [70] 赵艳红, 刘海风, 张弓木. PETN炸药爆轰产物状态方程的理论研究 [J]. 高压物理学报, 2009, 23(2): 143–149. doi: 10.3969/j.issn.1000-5773.2009.02.011ZHAO Y H, LIU H F, ZHANG G M. Equation of state of detonation products for PETN explosive [J]. Chinese Journal of High Pressure Physics, 2009, 23(2): 143–149. doi: 10.3969/j.issn.1000-5773.2009.02.011 [71] 赵艳红, 刘海风, 张广财. PBX9502炸药爆轰产物的状态方程 [J]. 爆炸与冲击, 2010, 30(6): 647–651.ZHAO Y H, LIU H F, ZHANG G C. Equation of state of detonation products for PBX9502 explosive [J]. Explosion and Shock Waves, 2010, 30(6): 647–651. [72] LÜ L, ZHANG L, YANG M L. Understanding the phase separation of N2/H2O and CO2/H2O binary systems through reactive force fields-based molecular dynamics simulations [J]. Journal of Applied Physics, 2018, 124: 235901. doi: 10.1063/1.5066585 [73] MENIKOFF R. Compaction wave profiles in granular HMX [J]. AIP Conference Proceedings, 2002, 620: 979–982. doi: 10.1063/1.1483701 [74] MENIKOFF R. Pore collapse and hot spots in HMX [J]. AIP Conference Proceedings, 2004, 706: 393–396. doi: 10.1063/1.1780261 [75] AUSTIN R A, BARTON N R, HOWARD W M, et al. Modeling pore collapse and chemical reactions in shock-loaded HMX crystals [J]. Journal of Physics: Conference Series, 2014, 500: 052002. doi: 10.1088/1742-6596/500/5/052002 [76] AUSTIN R A, BARTON N R, REAUGH J E, et al. Direct numerical simulation of shear localization and decomposition reactions in shock-loaded HMX crystal [J]. Journal of Applied Physics, 2015, 117: 185902. doi: 10.1063/1.4918538 [77] SPRINGER H K, TARVER C M, BASTEA S. Effects of high shock pressures and pore morphology on hot spot mechanisms in HMX [J]. AIP Conference Proceedings, 2017, 1793: 080002. doi: 10.1063/1.4971608 [78] ZHOU T T, LOU J F, ZHANG Y G, et al. Hot spot formation and chemical reaction initiation in shocked HMX crystals with nanovoids: a large-scale reactive molecular dynamics study [J]. Physical Chemistry Chemical Physics, 2016, 18(26): 17627–17645. doi: 10.1039/C6CP02015A [79] LONG Y, CHEN J. An investigation of the hot spot formation mechanism for energetic material [J]. Journal of Applied Physics, 2017, 122: 175105. doi: 10.1063/1.4996385 [80] LONG Y, CHEN J. A molecular dynamics study of the early-time mechanical heating in shock-loaded octahydro-1, 3, 5, 7-tetranitro-1, 3, 5, 7- tetrazocine-based explosives [J]. Journal of Applied Physics, 2014, 116: 033516. doi: 10.1063/1.4890715 [81] LONG Y, CHEN J. Theoretical study of the defect evolution for molecular crystal under shock loading [J]. Journal of Applied Physics, 2019, 125: 065107. doi: 10.1063/1.5067284 [82] ZHANG C Y. Computational investigation on the desensitizing mechanism of graphite in explosives versus mechanical stimuli: compression and glide [J]. The Journal of Physical Chemistry B, 2007, 111(22): 6208–6213. doi: 10.1021/jp070918d [83] ZHANG C Y. Understanding the desensitizing mechanism of olefin in explosives versus external mechanical stimuli [J]. The Journal of Physical Chemistry C, 2010, 114(11): 5068–5072. doi: 10.1021/jp910883x [84] LONG Y, LIU Y G, NIE F D, et al. Theoretical study of impacting and desensitizing for HMX-graphite mixture explosive [J]. Shock Waves, 2012, 22(6): 605–614. doi: 10.1007/s00193-012-0394-7 -

 下载:
下载:







 点击查看大图
点击查看大图
计量
- 文章访问数: 8297
- HTML全文浏览量: 2632
- PDF下载量: 62
