




New Developments of Time-Dependent Density Functional Theory and Its Applications
-
摘要: 密度泛函理论在材料计算研究领域得到了广泛的应用,然而它无法处理含时问题和材料的激发态性质。Runge-Gross定理奠定了含时密度泛函理论的基础,为研究这两类问题提供了有效的手段。经过三十多年的发展,含时密度泛函已被应用到量子化学、材料计算等多个领域,人们也更加了解其优势和不足。目前,含时密度泛函理论和方法仍在迅速发展。本文简要回顾含时密度泛函方法的发展历史,介绍近年来含时密度泛函在理论和应用方面的一些重要进展,总结当前在含时密度泛函领域存在的重要难题以及面临的挑战,展望其发展方向和趋势。Abstract: Nowadays density functional theory which was introduced in the mid-1960s has wide applications in material simulations. However, it is not able to deal with time-dependent problems and excited properties of materials. Time-dependent density functional theory (TDDFT) based on Runge-Gross theorem, provides a viable way to deal with these two problems. After thirty years’ development, TDDFT has been widely applied to many fields, such as quantum chemistry and material simulation, and our understanding of its advantages and weaknesses also grows. To date, TDDFT theory and method still develop rapidly. Here a brief historical review of TDDFT is first introduced. Then it is followed by a discussion of recent important developments on theory and applications of TDDFT. Finally we summarize some important problems and challenges that TDDFT are facing and attempt to offer some thoughts about where TDDFT will be progressing.
-
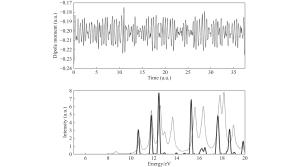


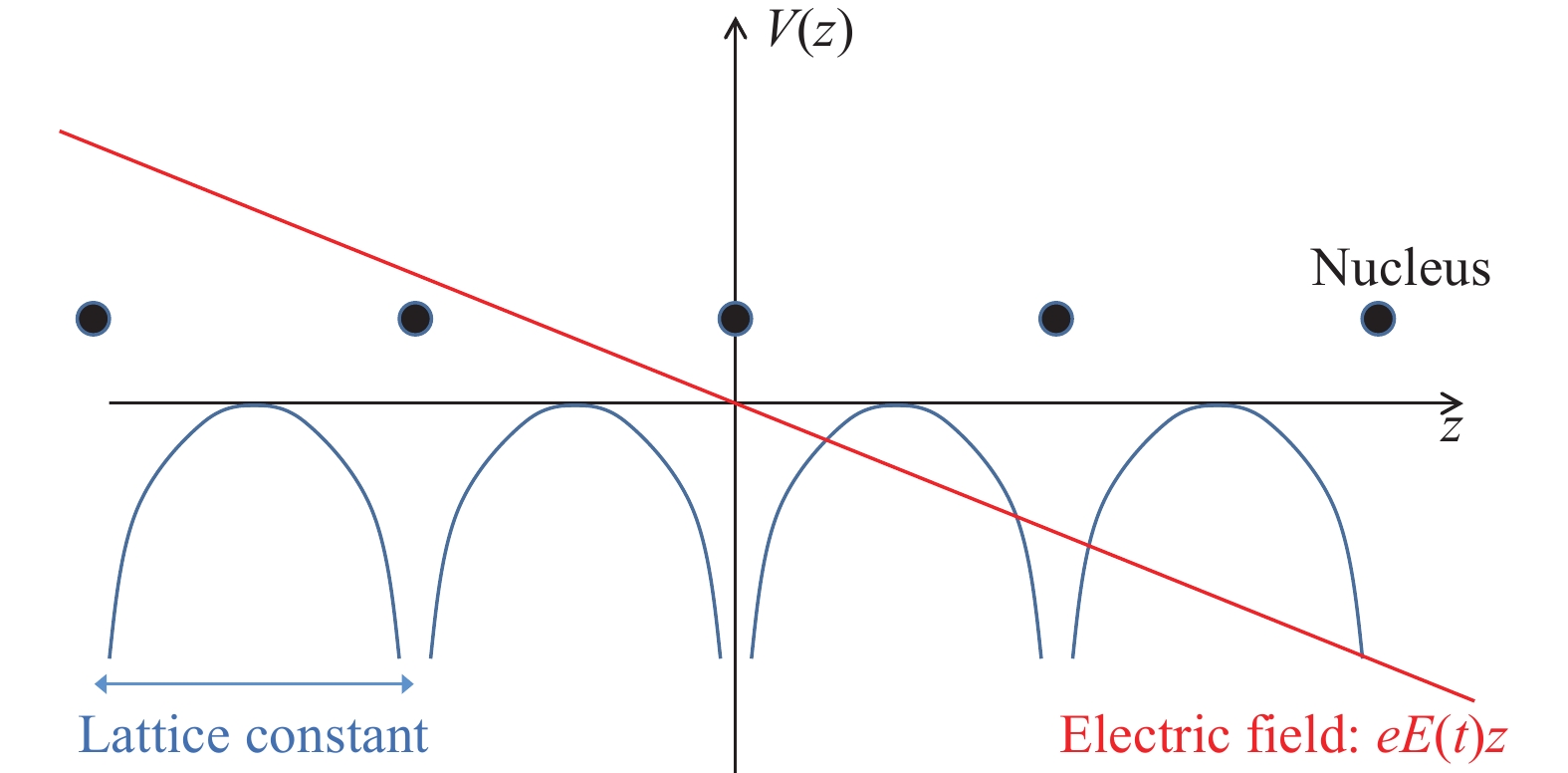
图 3 偶极近似破坏了晶体哈密顿量的空间周期性
Figure 3. Spatial periodicity of the Hamiltonian destroyed by the dipole approximation




表 1 一些气相Ar-TCNE体系的激发能E(单位eV)和振动强度f的计算和实验对比[57]
Table 1. Excitation energies E (eV) and oscillator strengths f of several gas phase Ar-TCNE systems: theory and experiment[57]
Ar B3LYP BNL ($\gamma $=0.5) BNL $\gamma^* $ Exp.[27] E f E $\gamma^* $ E f E f Benzene 2.1 0.03 4.4 0.33 3.8 0.03 3.59 0.02 Toluene 1.8 0.04 4.0 0.32 3.4 0.03 3.36 0.03 O-xylene 1.5 0 3.7 0.31 3.0 0.01 3.15 0.05 Naphthalene 0.9 0 3.3 0.32 2.7 0 2.60 0.01  下载: 导出CSV
下载: 导出CSV
-
[1] DIRAC P A M. Quantum mechanics of many-electron systems [J]. Proceedings of the Royal Society of London Series A, 1929, 123(792): 714–733. doi: 10.1098/rspa.1929.0094 [2] HOHENBERG P, KOHN W. Inhomogeneous electron gas [J]. Physical Review, 1964, 136(3B): B864–B871. doi: 10.1103/PhysRev.136.B864 [3] KOHN W, SHAM L J. Self-consistent equations including exchange and correlation effects [J]. Physical Review, 1965, 140(4A): A1133–A1138. doi: 10.1103/PhysRev.140.A1133 [4] BLOCH F. Bremsvermögen von Atomen mit mehreren Elektronen [J]. Zeitschrift für Physik A Hadrons and Nuclei, 1933, 81(5): 363–376. [5] PEUCKERT V. A new approximation method for electron systems [J]. Journal of Physics C: Solid State Physics, 1978, 11(24): 4945–4956. doi: 10.1088/0022-3719/11/24/023 [6] ZANGWILL A, SOVEN P. Density-functional approach to local-field effects in finite systems: photoabsorption in the rare gases [J]. Physical Review A, 1980, 21(5): 1561. doi: 10.1103/PhysRevA.21.1561 [7] DEB B M, GHOSH S K. Schrödinger fluid dynamics of many-electron systems in a time-dependent density-functional framework [J]. The Journal of Chemical Physics, 1982, 77(1): 342–348. doi: 10.1063/1.443611 [8] GHOSH S K, DEB B M. Dynamic polarizability of many-electron systems within a time-dependent density-functional theory [J]. Chemical Physics, 1982, 71(2): 295–306. doi: 10.1016/0301-0104(82)87030-4 [9] GHOSH S K, DEB B M. A density-functional calculation of dynamic dipole polarizabilities of noble gas atoms [J]. Theoretica Chimica Acta, 1983, 62(3): 209–217. doi: 10.1007/BF00548835 [10] GHOSH S K, DEB B M. A simple density-functional calculation of frequency-dependent multipole polarizabilities of noble gas atoms [J]. Journal of Molecular Structure: THEOCHEM, 1983, 103: 163–176. doi: 10.1016/0166-1280(83)85017-9 [11] BARTOLOTTI L J. Time-dependent extension of the Hohenberg-Kohn-Levy energy-density functional [J]. Physical Review A, 1981, 24(4): 1661–1667. doi: 10.1103/PhysRevA.24.1661 [12] BARTOLOTTI L J. Time-dependent Kohn-Sham density-functional theory [J]. Physical Review A, 1982, 26(4): 2243–2244. doi: 10.1103/PhysRevA.26.2243 [13] BARTOLOTTI L J. Variation-perturbation theory within a time-dependent Kohn-Sham formalism: an application to the determination of multipole polarizabilities, spectral sums, and dispersion coefficients [J]. The Journal of Chemical Physics, 1984, 80(11): 5687–5695. doi: 10.1063/1.446637 [14] BARTOLOTTI L J. Velocity form of the Kohn-Sham frequency-dependent polarizability equations [J]. Physical Review A, 1987, 36(9): 4492–4493. doi: 10.1103/PhysRevA.36.4492 [15] RUNGE E, GROSS E K U. Density-functional theory for time-dependent systems [J]. Physical Review Letters, 1984, 52(12): 997–1000. doi: 10.1103/PhysRevLett.52.997 [16] LEVY M. Universal variational functionals of electron densities, first-order density matrices, and natural spin-orbitals and solution of the v-representability problem [J]. Proceedings of the National Academy of Sciences, 1979, 76(12): 6062–6065. doi: 10.1073/pnas.76.12.6062 [17] LIEB E H. Density functionals for Coulomb systems [J]. International Journal of Quantum Chemistry, 1983, 24: 243–277. doi: 10.1002/(ISSN)1097-461X [18] VAN LEEUWEN R. Mapping from densities to potentials in time-dependent density-functional theory [J]. Physical Review Letters, 1999, 82(19): 3863–3866. doi: 10.1103/PhysRevLett.82.3863 [19] RUGGENTHALER M, GIESBERTZ K J H, PENZ M, et al. Density-potential mappings in quantum dynamics [J]. Physical Review A, 2012, 85(5): 052504. doi: 10.1103/PhysRevA.85.052504 [20] RUGGENTHALER M, VAN LEEUWEN R. Global fixed-point proof of time-dependent density-functional theory [J]. EPL (Europhysics Letters), 2011, 95(1): 13001. doi: 10.1209/0295-5075/95/13001 [21] ULLRICH C A. Time-dependent density-functional theory: concepts and applications [M]. OUP Oxford, 2011. [22] GIULIANI G, VIGNALE G. Quantum theory of the electron liquid [M]. Cambridge: Cambridge University Press, 2005. [23] ULLRICH C A, YANG Z. A brief compendium of time-dependent density functional theory [J]. Brazilian Journal of Physics, 2014, 44(1): 154–188. doi: 10.1007/s13538-013-0141-2 [24] GROSS E K U, KOHN W. Local density-functional theory of frequency-dependent linear response [J]. Physical Review Letters, 1985, 55(26): 2850–2852. doi: 10.1103/PhysRevLett.55.2850 [25] CASIDA M E. Time-dependent density functional response theory for molecules [M]//Recent Advances in Computational Chemistry. World Scientific, 1995: 155–192. [26] HIRATA S, HEAD-GORDON M. Time-dependent density functional theory within the Tamm-Dancoff approximation [J]. Chemical Physics Letters, 1999, 314(3/4): 291–299. [27] CASTRO A, MARQUES M A L, RUBIO A. Propagators for the time-dependent Kohn-Sham equations [J]. The Journal of Chemical Physics, 2004, 121(8): 3425–3433. doi: 10.1063/1.1774980 [28] BURNUS T, MARQUES M A L, GROSS E K U. Time-dependent electron localization function [J]. Physical Review A, 2005, 71(1): 010501. doi: 10.1103/PhysRevA.71.010501 [29] POHL A, REINHARD P G, SURAUD E. Towards single-particle spectroscopy of small metal clusters [J]. Physical Review Letters, 2000, 84(22): 5090–5093. doi: 10.1103/PhysRevLett.84.5090 [30] VÉNIARD V, TAIEB R, MAQUET A. Photoionization of atoms using time-dependent density functional theory [J]. Laser Physics, 2003, 13(4): 465–474. [31] DE GIOVANNINI U, VARSANO D, MARQUES M A L, et al. Ab initio angle-and energy-resolved photoelectron spectroscopy with time-dependent density-functional theory [J]. Physical Review A, 2012, 85(6): 062515. doi: 10.1103/PhysRevA.85.062515 [32] ROHRINGER N, PETER S, BURGDÖRFER J. Calculating state-to-state transition probabilities within time-dependent density-functional theory [J]. Physical Review A, 2006, 74(4): 042512. doi: 10.1103/PhysRevA.74.042512 [33] LI Y, ULLRICH C A. Time-dependent transition density matrix [J]. Chemical Physics, 2011, 391(1): 157–163. doi: 10.1016/j.chemphys.2011.02.001 [34] D’AGOSTA R, VIGNALE G. Non-V-representability of currents in time-dependent many-particle systems [J]. Physical Review B, 2005, 71(24): 245103. doi: 10.1103/PhysRevB.71.245103 [35] VIGNALE G. Mapping from current densities to vector potentials in time-dependent current density functional theory [J]. Physical Review B, 2004, 70(20): 201102. doi: 10.1103/PhysRevB.70.201102 [36] VIGNALE G, KOHN W. Current-dependent exchange-correlation potential for dynamical linear response theory [J]. Physical Review Letters, 1996, 77(10): 2037–2040. doi: 10.1103/PhysRevLett.77.2037 [37] VIGNALE G, ULLRICH C A, CONTI S. Time-dependent density functional theory beyond the adiabatic local density approximation [J]. Physical Review Letters, 1997, 79(24): 4878–4881. doi: 10.1103/PhysRevLett.79.4878 [38] MAITRA N T, SOUZA I, BURKE K. Current-density functional theory of the response of solids [J]. Physical Review B, 2003, 68(4): 045109. doi: 10.1103/PhysRevB.68.045109 [39] YABANA K, BERTSCH G F. Time-dependent local-density approximation in real time [J]. Physical Review B, 1996, 54(7): 4484–4487. [40] BERTSCH G F, IWATA J I, RUBIO A, et al. Real-space, real-time method for the dielectric function [J]. Physical Review B, 2000, 62(12): 7998–8002. [41] YABANA K, SUGIYAMA T, SHINOHARA Y, et al. Time-dependent density functional theory for strong electromagnetic fields in crystalline solids [J]. Physical Review B, 2012, 85(4): 045134. [42] MARQUES M A L, CASTRO A, BERTSCH G F, et al. Octopus: a first-principles tool for excited electron–ion dynamics [J]. Computer Physics Communications, 2003, 151(1): 60–78. [43] CASTRO A, APPEL H, OLIVEIRA M, et al. Octopus: a tool for the application of time-dependent density functional theory [J]. Physica Status Solidi (B), 2006, 243(11): 2465–2488. [44] ANDRADE X, STRUBBE D, DE GIOVANNINI U, et al. Real-space grids and the Octopus code as tools for the development of new simulation approaches for electronic systems [J]. Physical Chemistry Chemical Physics, 2015, 17(47): 31371–31396. [45] DREUW A, HEAD-GORDON M. Failure of time-dependent density functional theory for long-range charge-transfer excited states: the zincbacteriochlorin-bacteriochlorin and bacteriochlorophyll-spheroidene complexes [J]. Journal of the American Chemical Society, 2004, 126(12): 4007–4016. doi: 10.1021/ja039556n [46] HIERINGER W, GÖRLING A. Failure of time-dependent density functional methods for excitations in spatially separated systems [J]. Chemical Physics Letters, 2006, 419(4/5/6): 557–562. [47] AUTSCHBACH J. Charge-transfer excitations and time-dependent density functional theory: problems and some proposed solutions [J]. ChemPhysChem, 2009, 10(11): 1757–1760. doi: 10.1002/cphc.v10:11 [48] CASIDA M E, GUTIERREZ F, GUAN J, et al. Charge-transfer correction for improved time-dependent local density approximation excited-state potential energy curves: analysis within the two-level model with illustration for H2 and LiH [J]. The Journal of Chemical Physics, 2000, 113(17): 7062–7071. doi: 10.1063/1.1313558 [49] HU C, SUGINO O, MIYAMOTO Y. Modified linear response for time-dependent density-functional theory: application to Rydberg and charge-transfer excitations [J]. Physical Review A, 2006, 74(3): 032508. doi: 10.1103/PhysRevA.74.032508 [50] HU C, SUGIN O. Average excitation energies from time-dependent density functional response theory [J]. Journal of Chemical Physics, 2007, 126: 074112. doi: 10.1063/1.2436887 [51] TAWADA Y, TSUNEDA T, YANAGISAWA S, et al. A long-range-corrected time-dependent density functional theory [J]. The Journal of Chemical Physics, 2004, 120(18): 8425–8433. doi: 10.1063/1.1688752 [52] TOKURA S, TSUNEDA T, HIRAO K. Long-range-corrected time-dependent density functional study on electronic spectra of five-membered ring compounds and free-base porphyrin [J]. Journal of Theoretical and Computational Chemistry, 2006, 5(4): 925–944. doi: 10.1142/S0219633606002684 [53] VYDROV O A, SCUSERIA G E. Assessment of a long-range corrected hybrid functional [J]. The Journal of Chemical Physics, 2006, 125(23): 234109. doi: 10.1063/1.2409292 [54] LIVSHITS E, BAER R. A well-tempered density functional theory of electrons in molecules [J]. Physical Chemistry Chemical Physics, 2007, 9(23): 2932–2941. doi: 10.1039/b617919c [55] PEACH M J G, TELLGREN E I, SAŁEK P, et al. Structural and electronic properties of polyacetylene and polyyne from hybrid and coulomb-attenuated density functionals [J]. The Journal of Physical Chemistry A, 2007, 111(46): 11930–11935. doi: 10.1021/jp0754839 [56] LIVSHITS E, BAER R. A density functional theory for symmetric radical cations from bonding to dissociation [J]. The Journal of Physical Chemistry A, 2008, 112(50): 12789–12791. doi: 10.1021/jp803606n [57] STEIN T, KRONIK L, BAER R. Reliable prediction of charge transfer excitations in molecular complexes using time-dependent density functional theory [J]. Journal of the American Chemical Society, 2009, 131(8): 2818–2820. doi: 10.1021/ja8087482 [58] MAITRA N T, ZHANG F, CAVE R J, et al. Double excitations within time-dependent density functional theory linear response [J]. The Journal of Chemical Physics, 2004, 120(13): 5932–5937. doi: 10.1063/1.1651060 [59] GRITSENKO O V, BAERENDS E J. Double excitation effect in non-adiabatic time-dependent density functional theory with an analytic construction of the exchange–correlation kernel in the common energy denominator approximation [J]. Physical Chemistry Chemical Physics, 2009, 11(22): 4640–4646. doi: 10.1039/b903123e [60] ROMANIELLO P, SANGALLI D, BERGER J A, et al. Double excitations in finite systems [J]. The Journal of Chemical Physics, 2009, 130(4): 044108. doi: 10.1063/1.3065669 [61] SANGALLI D, ROMANIELLO P, ONIDA G, et al. Double excitations in correlated systems: a many–body approach [J]. The Journal of Chemical Physics, 2011, 134(3): 034115. doi: 10.1063/1.3518705 [62] SÄKKINEN N, MANNINEN M, VAN LEEUWEN R. The Kadanoff–Baym approach to double excitations in finite systems [J]. New Journal of Physics, 2012, 14(1): 013032. doi: 10.1088/1367-2630/14/1/013032 [63] HUIX-ROTLLANT M, IPATOV A, RUBIO A, et al. Assessment of dressed time-dependent density-functional theory for the low-lying valence states of 28 organic chromophores [J]. Chemical Physics, 2011, 391(1): 120–129. doi: 10.1016/j.chemphys.2011.03.019 [64] FURCHE F, AHLRICHS R. Absolute configuration of D2-symmetric fullerene C84 [J]. Journal of the American Chemical Society, 2002, 124(15): 3804–3805. doi: 10.1021/ja012207d [65] LIU J, LIANG W Z. Molecular-orbital-free algorithm for the excited-state force in time-dependent density functional theory [J]. The Journal of Chemical Physics, 2011, 134(4): 044114. doi: 10.1063/1.3548063 [66] LEVINE B G, KO C, QUENNEVILLE J, et al. Conical intersections and double excitations in time-dependent density functional theory [J]. Molecular Physics, 2006, 104(5/6/7): 1039–1051. [67] TAPAVICZA E, TAVERNELLI I, ROTHLISBERGER U, et al. Mixed time-dependent density-functional theory/classical trajectory surface hopping study of oxirane photochemistry [J]. The Journal of Chemical Physics, 2008, 129(12): 124108. doi: 10.1063/1.2978380 [68] KADUK B, VAN VOORHIS T. Communication: conical intersections using constrained density functional theory–configuration interaction [J]. Journal of Chemical Physics, 2010, 133: 061102. doi: 10.1063/1.3470106 [69] MAITRA N T. On correlated electron-nuclear dynamics using time-dependent density functional theory [J]. The Journal of Chemical Physics, 2006, 125(1): 014110. doi: 10.1063/1.2210471 [70] GONZE X, GHOSEZ P, GODBY R W. Density-polarization functional theory of the response of a periodic insulating solid to an electric field [J]. Physical Review Letters, 1995, 74(20): 4035. doi: 10.1103/PhysRevLett.74.4035 [71] GHOSEZ P, GONZE X, GODBY R W. Long-wavelength behavior of the exchange-correlation kernel in the Kohn-Sham theory of periodic systems [J]. Physical Review B, 1997, 56(20): 12811–12817. doi: 10.1103/PhysRevB.56.12811 [72] ONIDA G, REINING L, RUBIO A. Electronic excitations: density-functional versus many-body Green’s-function approaches [J]. Reviews of Modern Physics, 2002, 74(2): 601–659. doi: 10.1103/RevModPhys.74.601 [73] BOTTI S, SOTTILE F, VAST N, et al. Long-range contribution to the exchange-correlation kernel of time-dependent density functional theory [J]. Physical Review B, 2004, 69(15): 155112. doi: 10.1103/PhysRevB.69.155112 [74] SHARMA S, DEWHURST J K, SANNA A, et al. Bootstrap approximation for the exchange-correlation kernel of time-dependent density-functional theory [J]. Physical Review Letters, 2011, 107(18): 186401. doi: 10.1103/PhysRevLett.107.186401 [75] SHARMA S, DEWHURST J K, SANNA A, et al. Enhanced excitonic effects in the energy loss spectra of LiF and Ar at large momentum transfer [J]. New Journal of Physics, 2012, 14(5): 053052. doi: 10.1088/1367-2630/14/5/053052 [76] TANCOGNE-DEJEAN N, MÜCKE O D, KÄRTNER F X, et al. Impact of the electronic band structure in high-harmonic generation spectra of solids [J]. Physical Review Letters, 2017, 118(8): 087403. doi: 10.1103/PhysRevLett.118.087403 [77] BACZEWSKI A D, SHULENBURGER L, DESJARLAIS M P, et al. X-ray thomson scattering in warm dense matter without the chihara decomposition [J]. Physical Review Letters, 2016, 116(11): 115004. doi: 10.1103/PhysRevLett.116.115004 [78] MO C, FU Z, KANG W, et al. First-principles estimation of electronic temperature from X-ray thomson scattering spectrum of isochorically heated warm dense matter [J]. Physical Review Letters, 2018, 120(20): 205002. doi: 10.1103/PhysRevLett.120.205002 [79] SPERLING P, GAMBOA E J, LEE H J, et al. Free-electron X-ray laser measurements of collisional-damped plasmons in isochorically heated warm dense matter [J]. Physical Review Letters, 2015, 115(11): 115001. doi: 10.1103/PhysRevLett.115.115001 [80] RAMAKRISHNAN R, HARTMANN M, TAPAVICZA E, et al. Electronic spectra from TDDFT and machine learning in chemical space [J]. The Journal of Chemical Physics, 2015, 143(8): 084111. doi: 10.1063/1.4928757 [81] FUKS J I, NIELSEN S E B, RUGGENTHALER M, et al. Time-dependent density functional theory beyond Kohn–Sham Slater determinants [J]. Physical Chemistry Chemical Physics, 2016, 18(31): 20976–20985. doi: 10.1039/C6CP00722H [82] NIELSEN S E B, RUGGENTHALER M, VAN LEEUWEN R. Many-body quantum dynamics from the density [J]. EPL (Europhysics Letters), 2013, 101(3): 33001. doi: 10.1209/0295-5075/101/33001 [83] ELLIOTT P, MAITRA N T. Propagation of initially excited states in time-dependent density-functional theory [J]. Physical Review A, 2012, 85(5): 052510. doi: 10.1103/PhysRevA.85.052510 [84] CASTRO A, MARQUES M A L, ALONSO J A, et al. Excited states dynamics in time-dependent density functional theory [J]. The European Physical Journal D, 2004, 28(2): 211–218. doi: 10.1140/epjd/e2003-00306-3 [85] ALONSO J L, ANDRADE X, ECHENIQUE P, et al. Efficient formalism for large-scale ab initio molecular dynamics based on time-dependent density functional theory [J]. Physical Review Letters, 2008, 101(9): 096403. doi: 10.1103/PhysRevLett.101.096403 [86] MENG S, KAXIRAS E. Real-time, local basis-set implementation of time-dependent density functional theory for excited state dynamics simulations [J]. The Journal of Chemical Physics, 2008, 129(5): 054110. doi: 10.1063/1.2960628 [87] MENG S, KAXIRAS E. Mechanisms for ultrafast nonradiative relaxation in electronically excited eumelanin constituents [J]. Biophysical Journal, 2008, 95(9): 4396–4402. doi: 10.1529/biophysj.108.135756 [88] TULLY J C. Molecular dynamics with electronic transitions [J]. The Journal of Chemical Physics, 1990, 93(2): 1061–1071. doi: 10.1063/1.459170 [89] TAPAVICZA E, TAVERNELLI I, ROTHLISBERGER U. Trajectory surface hopping within linear response time-dependent density-functional theory [J]. Physical Review Letters, 2007, 98(2): 023001. doi: 10.1103/PhysRevLett.98.023001 [90] TAVERNELLI I, CURCHOD B F E, ROTHLISBERGER U. Mixed quantum-classical dynamics with time-dependent external fields: a time-dependent density-functional-theory approach [J]. Physical Review A, 2010, 81(5): 052508. doi: 10.1103/PhysRevA.81.052508 [91] KRISHTAL A, CERESOLI D, PAVANELLO M. Subsystem real-time time dependent density functional theory [J]. The Journal of Chemical Physics, 2015, 142(15): 154116. doi: 10.1063/1.4918276 [92] PAVANELLO M. On the subsystem formulation of linear-response time-dependent DFT [J]. The Journal of Chemical Physics, 2013, 138(20): 204118. doi: 10.1063/1.4807059 [93] NEUGEBAUER J. Couplings between electronic transitions in a subsystem formulation of time-dependent density functional theory [J]. The Journal of Chemical Physics, 2007, 126(13): 134116. doi: 10.1063/1.2713754 -

 下载:
下载:





 点击查看大图
点击查看大图
图(7) / 表(1)
计量
- 文章访问数: 10270
- HTML全文浏览量: 3985
- PDF下载量: 117
