




Computation and Simulation of High-Pressure Properties of Complex Materials: A Brief Review on the Methods Based on First-Principles
-
摘要: 对基于第一性原理量子力学计算与模拟在复杂材料体系高压性质研究中的应用进行了简要回顾与综述,重点介绍了在合金、含缺陷材料以及电子强关联材料等复杂体系研究中的部分应用,并讨论了将量子力学原理与基于集团展开法、格子气模型、准模拟退火等物理模型相结合而发展出的一系列计算方法的优势与不足。本文所涵盖的内容仅仅是第一性原理计算方法从简单体系向复杂体系发展中的一小部分,但都具有一定的代表性,希望对发展更先进高效的具有预测能力的多尺度方法提供有益的参考。Abstract: This work briefly summarizes and reviewes the first-principles quantum mechanics calculations and simulations on the high-pressure properties of complex materials. We emphasized the applications in alloys and intermetallic compounds, materials with defects and strongly correlated electron systems. A series of methods, including cluster expansion method, lattice gas model, and quasi-annealing simulation approach, have been developed by combing quantum mechanics calculations with the statistical mechanics principles. Their pros and cons are discussed. The contents covered in this review are just a small portion of the first-principles methods that are evolving to tackle the complex systems. But they are of representative, and a retrospect of them might be helpful for developing better methods with high efficiency and good predictability for multiple-scale simulations.
-
Key words:
- first-principles method /
- complex system /
- high pressure /
- quantum mechanics /
- density functional theory
-

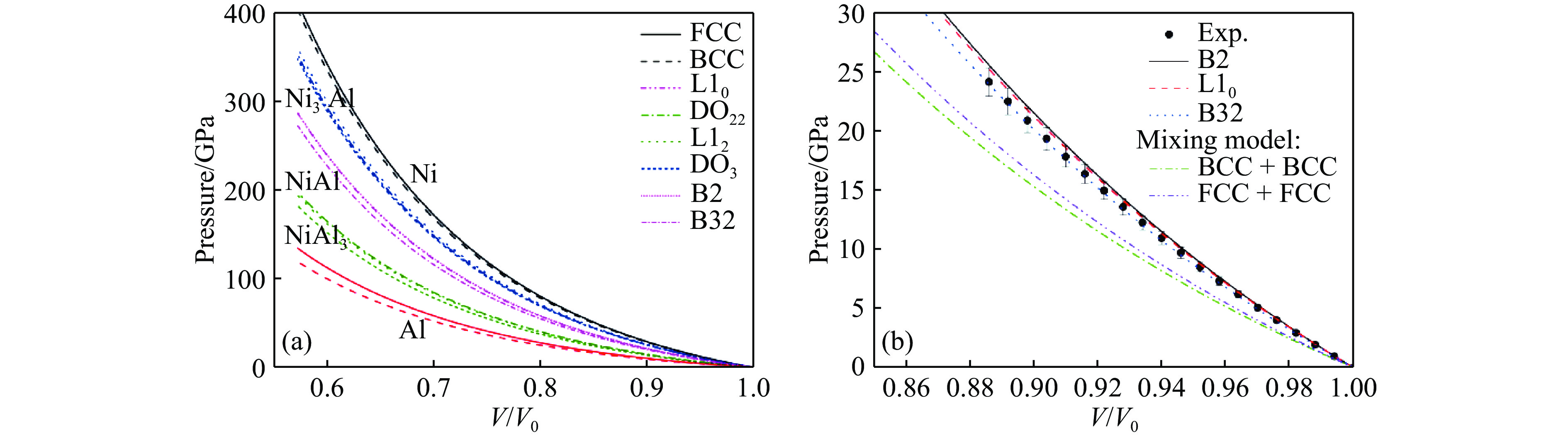
图 1 基于FCC和BCC晶格的Ni-Al合金第一性原理压缩曲线(a)以及Ni-Al合金有序相的计算压缩曲线、实验测量[38]和混合物模型结果[41](b)
Figure 1. The first-principle calculated compression curves of Ni-Al alloys based on FCC and BCC lattices (a); The calculated compression curves of ordered phases of NiAl alloys, the experimental measurements[38] and the results of the mixture model[41] (b)
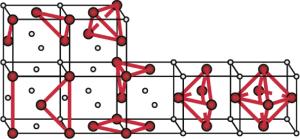
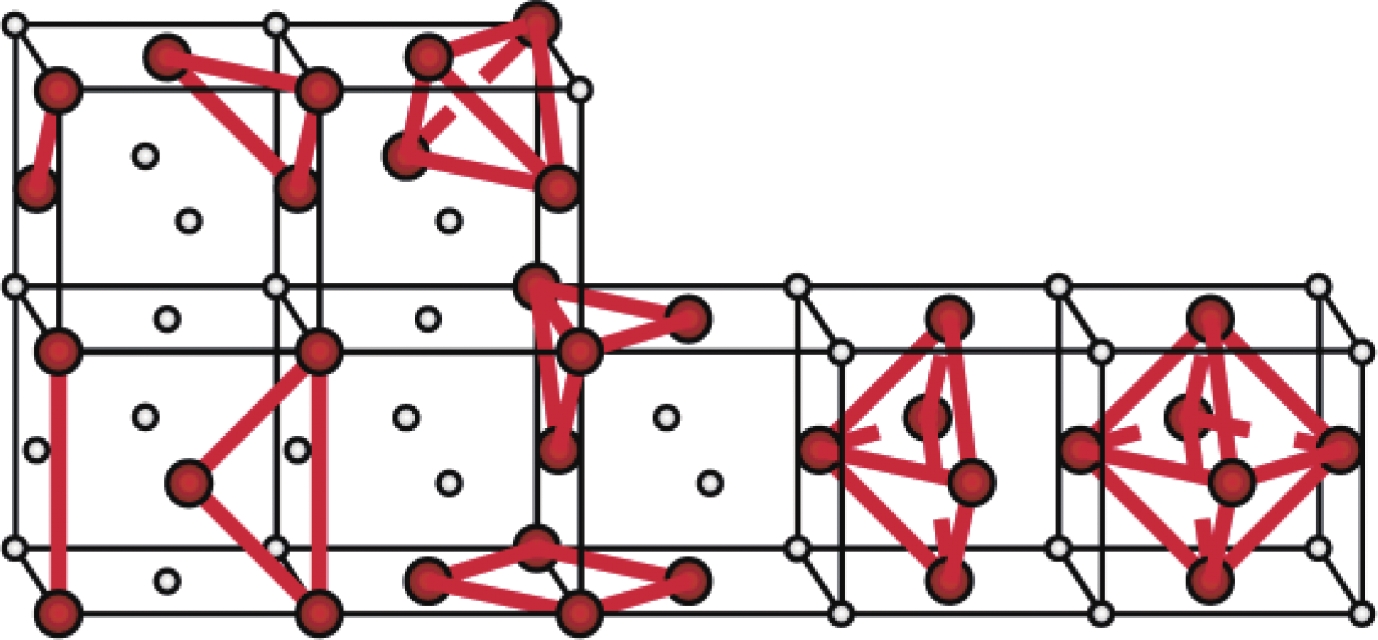
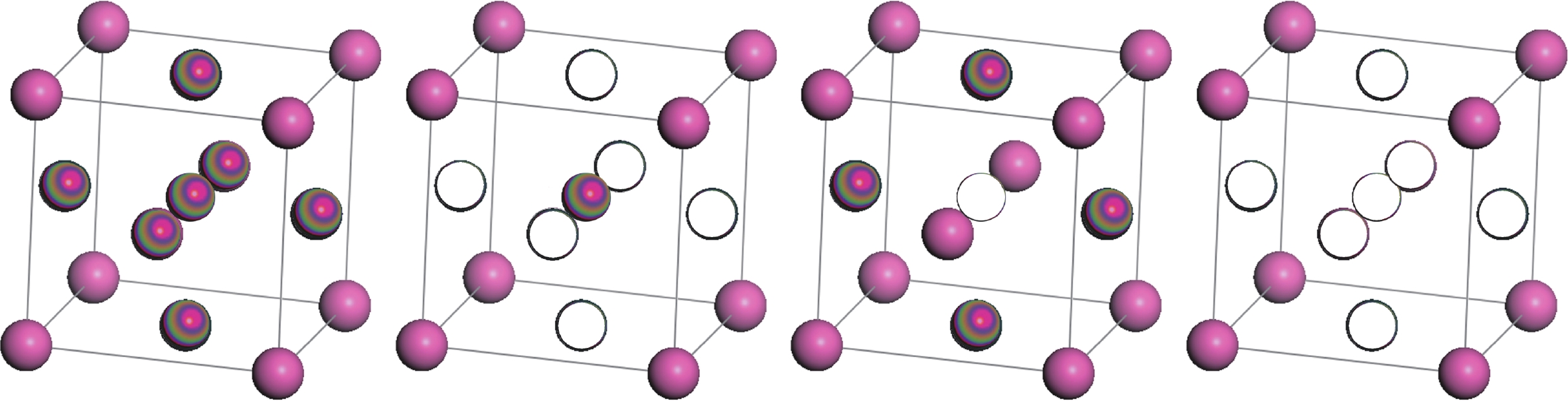
图 2 FCC晶格上的四面体展开和四面体-八面体展开集团示意图
Figure 2. Tetrahedron expansion and tetrahedron-octahedron expansion clusters on FCC lattice
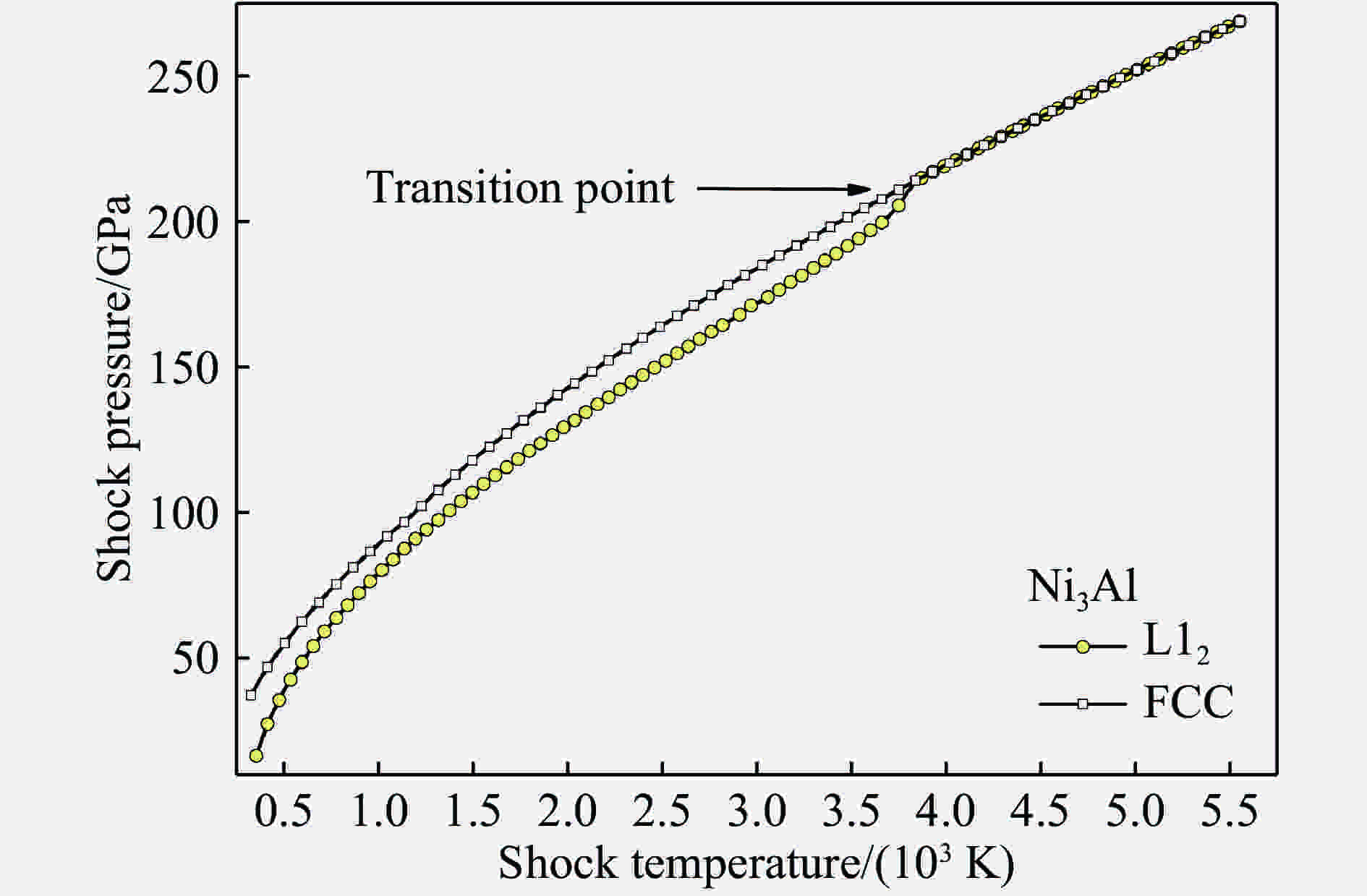
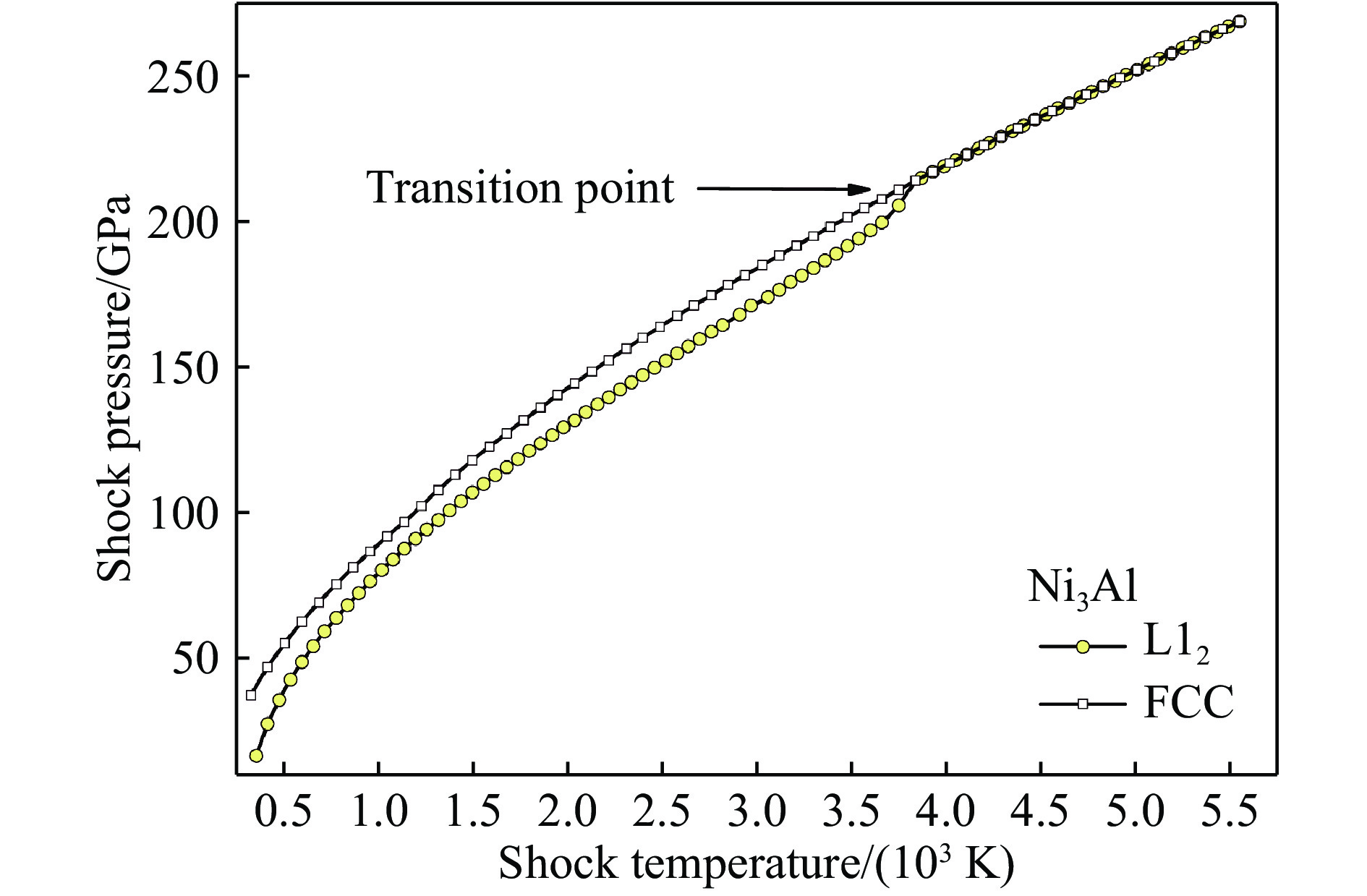
图 3 理论预测的Ni3Al中有序-无序相变导致的冲击绝热线跃变
Figure 3. Theoretically predicted Hugoniot kink caused by ordered-disordered phase transition in Ni3Al
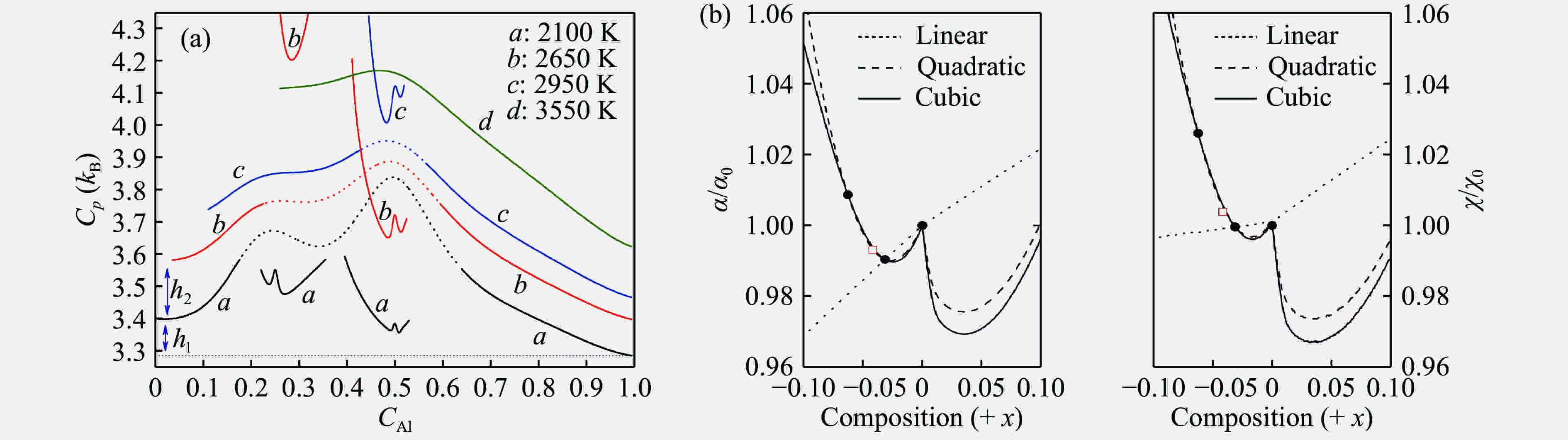
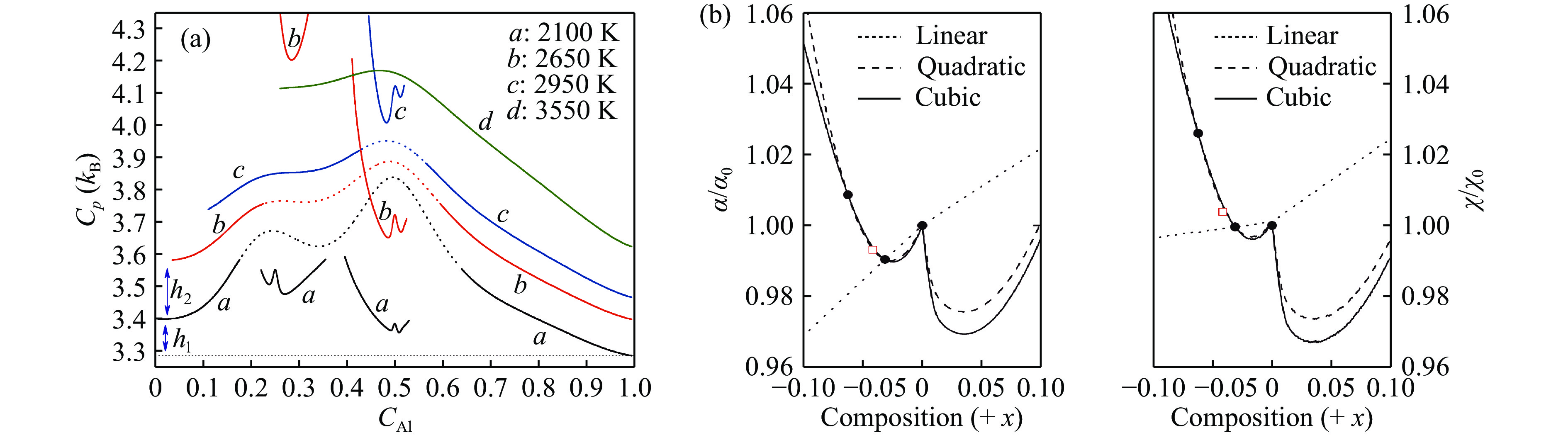
图 4 30 GPa压力下Ni-Al合金的定压热容随组分和温度的变化[61](a);UO2中点缺陷导致的热胀系数
${\alpha}$ 和压缩系数$ {\chi}$ 随标准化学比偏离的“W”形变化[64](b)Figure 4. The constant pressure heat capacity of Ni-Al alloy varies with composition and temperature at 30 GPa[61] (a); the "W" shape curve of the thermal expansion coefficient
${\alpha}$ and compression coefficient$ {\chi}$ caused by point defect in UO2 as a function of the deviation from the stoichiometry[64] (b) -
[1] SCHRÖDINGER E. An undulatory theory of the mechanics of atoms and molecules [J]. Physical Review, 1926, 28(6): 1049–1070. doi: 10.1103/PhysRev.28.1049 [2] HEISENBERG W. Quantum-theoretical re-interpretation of kinematic and mechanical relations [J]. Zeitschrift für Physik, 1925, 33: 879. doi: 10.1007/BF01328377 [3] DIRAC P A M. The quantum theory of the electron [J]. Proceedings of the Royal Society of London, 1928, 117(778): 610. doi: 10.1098/rspa.1928.0023 [4] PAULI W. On the connection of the arrangement of electron groups in atoms with the complex structure of spectra [J]. Zeitschrift für Physik, 1925, 31: 765–765. doi: 10.1007/BF02980631 [5] HARTREE D R. The wave mechanics of an atom with a non-coulomb central field [J]. Mathematical Proceedings of the Cambridge, 1928, 24: 89. doi: 10.1017/S0305004100011919 [6] FOCK F. Nherungs methode zur lsung des quanten mechanischen mehrkrper problems [J]. Zeitschrift für Physik, 1930, 61: 126. doi: 10.1007/BF01340294 [7] NITYANANDA, HOHENBERG P, KOHN W. Inhomogeneous electron gas [J]. Resonance, 1964, 136: B864. [8] KOHN W, SHAM L J. Self-consistent equations including exchange and correlation effects [J]. Physical Review, 1965, 140: A1133. doi: 10.1103/PhysRev.140.A1133 [9] KOHN W. Nobel lecture: electronic structure of matter-wave functions and density functionals [J]. Reviews of Modern Physics, 1999, 71(5): 1253. doi: 10.1103/RevModPhys.71.1253 [10] PERDEW J P, CHEVARY J A, VOSKO S H, et al. Atoms, molecules, solids, and surfaces: applications of the generalized gradient approximation for exchange and correlation [J]. Physical Review B, 1992, 46(11): 6671. doi: 10.1103/PhysRevB.46.6671 [11] SUN J, MARSMAN M, CSONKA G, et al. Self-consistent meta-generalized gradient approximation within the projector-augmented-wave method [J]. Physical Review B, 2011, 84(3): 035117. doi: 10.1103/PhysRevB.84.035117 [12] ZHAO Y, TRUHLAR D G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions [J]. Journal of Chemical Physics, 2006, 125(19): 194101. doi: 10.1063/1.2370993 [13] DION M, RYDBERG H, SCHRODER E, et al. Van der Waals density functional for general geometries [J]. Physical Review Letters, 2004, 92(24): 246401. doi: 10.1103/PhysRevLett.92.246401 [14] PAIER J, HIRSCHL R, MARSMAN M, et al. The Perdew-Burke-Ernzerhof exchange-correlation functional applied to the G2-1 test set using a plane-wave basis set [J]. The Journal of Chemical Physics, 2005, 122(23): 234102. doi: 10.1063/1.1926272 [15] LEE C, YANG W, PARR R G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density [J]. Physical Review B, 1988, 37(2): 786–789. [16] BECKE A D. Density-functional exchange-energy approximation with correct asymptotic behavior [J]. Physical Review A, 1988, 38: 3098. doi: 10.1103/PhysRevA.38.3098 [17] BECKE A D. Correlation energy of an inhomogeneous electron gas: a coordinate-space model [J]. The Journal of Chemical Physics, 1988, 88(2): 1053–1062. doi: 10.1063/1.454274 [18] LIECHTENSTEIN A I, ANISIMOV V I, ZAANE J. Density-functional theory and strong interactions: orbital ordering in Mott-Hubbard insulators [J]. Physical Review B, 1995, 52(8): R5467. doi: 10.1103/PhysRevB.52.R5467 [19] DUDAREV S L, BOTTON G A, SAVRASOV S Y, et al. Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA+U study [J]. Physical Review B, 1998, 57(3): 1505. doi: 10.1103/PhysRevB.57.1505 [20] SHISHKIN M, KRESSE G. Implementation and performance of frequency-dependent GW method within PAW framework [J]. Physical Review B, 2006, 74(3): 035101. [21] SHISHKIN M, KRESSE G. Self-consistent GW calculations for semiconductors and insulators [J]. Physical Review B, 2007, 75(23): 235102. doi: 10.1103/PhysRevB.75.235102 [22] FUCHS F, FURTHMULLER J, BECHSTEDT F, et al. Quasiparticle band structure based on a generalized Kohn-Sham scheme [J]. Physical Review B, 2007, 76: 115109. doi: 10.1103/PhysRevB.76.115109 [23] HARL J, KRESSE G. Accurate bulk properties from approximate many-body techniques [J]. Physical Review Letters, 2009, 103(5): 056401. doi: 10.1103/PhysRevLett.103.056401 [24] HARL J, SCHIMKA L, KRESSE G. Assessing the quality of the random phase approximation for lattice constants and atomization energies of solids [J]. Physical Review B, 2010, 50(11): 115126. [25] BOETTGER J C, TRICKEY S B. Equation of state and properties of lithium [J]. Physical Review B, 1985, 32(6): 3391–3398. doi: 10.1103/PhysRevB.32.3391 [26] GENG H Y, SONG H X, LI J F, et al. High-pressure behavior of dense hydrogen up to 3.5 TPa from density functional theory calculations [J]. Journal of Applied Physics, 2012, 111: 063510. doi: 10.1063/1.3694793 [27] ALFE D, PRICE C D, GILLAN M J. Thermodynamics of hexagonal-close-packed iron under Earth’s core conditions [J]. Physical Review B, 2001, 64(4): 045123. doi: 10.1103/PhysRevB.64.045123 [28] KRESS J D, MAZEVET S, COLLINS L A, et al. Density-functional calculation of the Hugoniot of shocked liquid nitrogen [J]. Physical Review B, 2000, 63: 024203. doi: 10.1103/PhysRevB.63.024203 [29] RECOULES V, MAZEVET S. Temperature and density dependence of XANES spectra in warm dense aluminum plasmas [J]. Physical Review B, 2009, 80: 064110. doi: 10.1103/PhysRevB.80.064110 [30] VAKS V G , KATSNELSON M I , KORESHKOV V G, et al. An experimental and theoretical study of martensitic phase transitions in Li and Na under pressure [J]. Journal of Physics Condensed Matter, 1989, 1: 5319–5335. doi: 10.1088/0953-8984/1/32/001 [31] MONSERRAT B, DRUMMOND N D, NEEDS R J. Anharmonic vibrational properties in periodic systems: energy, electron-phonon coupling, and stress [J]. Physical Review B, 2013, 87: 144302. doi: 10.1103/PhysRevB.87.144302 [32] GRABOWSKI B, ISMER L, HICKEL T, et al. Ab initio up to the melting point: anharmonicity and vacancies in aluminum [J]. Physical Review B, 2009, 79(13): 134106. doi: 10.1103/PhysRevB.79.134106 [33] SCHAEFFER A M J, TALMADGE W B, TEMPLE S R, et al. High pressure melting of lithium [J]. Physical Review Letters, 2012, 109: 185702. doi: 10.1103/PhysRevLett.109.185702 [34] LEPESHKINA S V, MAGNITSKAYAC M V, MAKSIMOVA E G. Lattice dynamics and melting features of Li and Na [J]. Jetp Letters, 2009, 89: 586–591. doi: 10.1134/S0021364009110137 [35] MUNEHISA M, QUAN Y, JUNYA O, et al. Multiple quantum phase transitions of plutonium compounds [J]. Life Sciences, 2011, 84(4): 041105. [36] NICOLA L, YAO Y X, WANG C Z, et al. Phase diagram and electronic structure of praseodymium and plutonium [J]. Physical Review X, 2015, 5(1): 011008. doi: 10.1103/PhysRevX.5.011008 [37] ANDRAULT D, BOLFAN-CASANOVA N, OHTAKA O, et al. Melting diagrams of Fe-rich alloys determined from synchrotron in situ measurements in the 15–23 GPa pressure range [J]. Physics of the Earth and Planetary Interiors, 2009, 174: 181–191. doi: 10.1016/j.pepi.2008.09.020 [38] MOUSAZADEHA M H, FARAMARZIB E, MALEKIA Z. Equation of state for thermodynamic properties of pure and mixtures liquid alkali metals [J]. Thermochimica Acta, 2010, 511: 147–151. doi: 10.1016/j.tca.2010.08.006 [39] OTTO J W, VASSILIOU J K, FROMMEYER G. Equation of state of polycrystalline Ni50Al50 [J]. Journal of Materials Research, 1997, 12(11): 3106. doi: 10.1557/JMR.1997.0405 [40] HIRAO N, OHTANI E, KONDO T, et al. Equation of state of iron-silicon alloys to megabar pressure [J]. Physics & Chemistry of Minerals, 2004, 31(6): 329–336. [41] GENG H Y, CHEN N X, SLUITER M H F. First-principles equation of state and phase stability for the Ni-Al system under high pressures [J]. Physical Review B, 2004, 70(9): 094203. doi: 10.1103/PhysRevB.70.094203 [42] KIKUCHI R. A theory of cooperative phenomena [J]. Physical Review, 1951, 81: 988. doi: 10.1103/PhysRev.81.988 [43] BARKER J A. Methods of approximation in the theory of regular mixtures [J]. Proceedings of the Royal Society A: Mathematical, 1952, 216: 45. [44] MORITA T. General structure of distribution function for the Heisenberg model and the Ising model [J]. Journal of Mathematical Physics, 1972, 13(1): 115–123. doi: 10.1063/1.1665840 [45] MORITA T. Cluster variation method and Mobius inversion formula [J]. Journal of Statistical Physics, 1990, 59: 819. doi: 10.1007/BF01025852 [46] SCHLIJPER A G. Exact variational methods and cluster-variation approximations [J]. Journal of Statistical Physics, 1984, 35(3/4): 285–301. [47] SCHLIJPER A G. Convergence of the cluster-variation method in the thermodynamic limit [J]. Physical Review B, 1983, 27(11): 6841. doi: 10.1103/PhysRevB.27.6841 [48] CACCIAMANI G, CHANG Y A, GRIMVALL G, et al. Thermodynamic modeling of solutions and alloys [J]. Calphad-Computer Coupling of Phase Diagrams & Thermochemistry, 1997, 21(2): 155–170. [49] HORIUCHI T, UZAWA H, IGARASHI M, et al. Determination of Lennard-Jones type potential for Fe-Pd phase [J]. Calphad, 2002, 26(1): 3–14. doi: 10.1016/S0364-5916(02)00021-4 [50] SANCHEZ J M, DUCASTELLE F, GRATIAS D. Generalized cluster description of multicomponent systems [J]. Physica A, 1984, 128: 334. doi: 10.1016/0378-4371(84)90096-7 [51] CONNOLLY J W D, WILLIAMS A R. Density-functional theory applied to phase transformations in transition-metal alloys [J]. Physical Review B, 1983, 27: 5169. doi: 10.1103/PhysRevB.27.5169 [52] GARBULSKY G D, CEDER G. Linear-programming method for obtaining effective cluster interactions in alloys from total-energy calculations: application to fcc Pd-V system [J]. Physical Review B, 1995, 51: 67. doi: 10.1103/PhysRevB.51.67 [53] GARBULSKY G D, CEDER G. Effect of lattice vibrations on the ordering tendencies in substitutional binary alloys [J]. Physical Review B, 1994, 49: 6327. doi: 10.1103/PhysRevB.49.6327 [54] VAN DE WALLE A, CEDER G, WAGHMARE U V. First-principles computation of the vibrational entropy of ordered and disordered Ni3Al [J]. Physical Review Letters, 1998, 80(22): 4911–4914. doi: 10.1103/PhysRevLett.80.4911 [55] VAN DE WALLE A, CEDER G. The effect of lattice vibrations on substitutional alloy thermodynamics [J]. Reviews of Modern Physics, 2002, 74(1): 11–45. doi: 10.1103/RevModPhys.74.11 [56] GENG H Y, SLUITER M H F, CHEN N X. Cluster expansion of electronic excitations: application to fcc Ni-Al alloys [J]. The Journal of Chemical Physics, 2005, 122: 214706. doi: 10.1063/1.1926276 [57] CEDER G, GARBULSKY G D, TEPESCH P D. Convergent real-space cluster expansion for configurational disorder in ionic systems [J]. Physical Review B, 1995, 51(21): 11257. [58] TOKAR V I. A new cluster method in lattice statistics [J]. Computational Materials Science, 1997, 8(1/2): 8–15. [59] NATALIYA P, BENOÎT A, JULIEN T, et al. A convex hull algorithm for a grid minimization of Gibbs energy as initial step in equilibrium calculations in two-phase multicomponent alloys [J]. Computational Materials Science, 2012, 61(11): 54–66. [60] SLUITER M H F, WATANABE Y, FONTAINE D, et al. First-principles calculation of the pressure dependence of phase equilibria in the Al-Li system [J]. Physical Review B, 1996, 53(11): 6137–6151. [61] GENG H Y, SLUITER M H F, CHEN N X. Order-disorder effects on the equation of state for fcc Ni-Al alloys [J]. Physical Review B, 2005, 72(1): 014204. doi: 10.1103/PhysRevB.72.014204 [62] SLUITER M, KAWAZOE Y. Explanation for the configurational heat capacity of ordered phases [J]. Physical Review B, 1999, 59(5): 3280. doi: 10.1103/PhysRevB.59.3280 [63] GENG H Y, CHEN N X, SLUITER M H F. Shock induced order-disorder transformation in Ni3Al [J]. Physical Review B, 2005, 71(1): 012105. doi: 10.1103/PhysRevB.71.012105 [64] GENG H Y, SONG H X, WU Q. Theoretical assessment on the possibility of constraining point-defect energetics by pseudo phase transition pressures [J]. Physical Review B, 2013, 87: 174107. doi: 10.1103/PhysRevB.87.174107 [65] GENG H Y, SLUITER M H F, CHEN N X. Hybrid cluster expansions for local structural relaxations [J]. Physical Review B, 2006, 73: 012202. doi: 10.1103/PhysRevB.73.012202 [66] 耿华运. 替位合金物态方程的有序-无序效应 [D]. 北京: 清华大学, 2005. [67] GENG H Y, CHEN Y, KANETA Y. Point defects and clustering in uranium dioxide by LSDA+U calculations [J]. Physical Review B, 2008, 77: 104120. doi: 10.1103/PhysRevB.77.104120 [68] SLUITER M H F. Lattice stability prediction of elemental tetrahedrally close-packed structures [J]. Acta Materialia, 2007, 55(11): 3707–3718. doi: 10.1016/j.actamat.2007.02.016 [69] GENG H Y, SONG HONG X, WU Q. Anomalies in nonstoichiometric uranium dioxide induced by a pseudo phase transition of point defects [J]. Physical Review B, 2012, 85(14): 144111. doi: 10.1103/PhysRevB.85.144111 [70] GENG H Y, SONG H X, JIN K, et al. First-principles study on oxidation effects in uranium oxides and high-pressure high-temperature behavior of point defects in uranium dioxide [J]. Physical Review B, 2011, 84(11): 174115. [71] GENG H Y, CHEN Y, KANETA Y, et al. Ab initio investigation on oxygen defect clusters in UO2+ x [J]. Applied Physics Letters, 2008, 93: 201903. doi: 10.1063/1.3035846 [72] GENG H Y, CHEN Y, KANETA Y, et al. Stability mechanism of cuboctahedral clusters in UO2+ x: first-principles calculations [J]. Physical Review B, 2008, 77: 180101. doi: 10.1103/PhysRevB.77.180101 [73] KRESSE G, FURTHMULLER J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set [J]. Physical Review B, 1996, 54(16): 11169–11186. doi: 10.1103/PhysRevB.54.11169 [74] GENG H Y, CHEN Y, KANETA Y, et al. Interplay of defect cluster and the stability of xenon in uranium dioxide from density functional calculations [J]. Physical Review B, 2010, 82(9): 094106. doi: 10.1103/PhysRevB.82.094106 [75] ANTOINE C, MARCO K, NICALS L, et al. GGA+U study of uranium mononitride: a comparison of the U-ramping and occupation matrix schemes and incorporation energies of fission products [J]. Journal of Nuclear Materials, 2016, 478(1): 119–124. [76] MEREDIG B, THOMPSON A, HANSEN H A, et al. Method for location low-energy solutions within DFT+U [J]. Physical Review B, 2010, 82(19): 195128. doi: 10.1103/PhysRevB.82.195128 [77] YI S, QING P, GANG L. Quantum mechanical modeling of hydrogen assisted cracking in aluminum [J]. Physical Review B, 2013, 88(10): 104109. doi: 10.1103/PhysRevB.88.104109 [78] CHEN Z, ZHANG X, LU G. Multiscale computational design of core/shell nanoparticles for oxygen reduction reaction [J]. The Journal of Physical Chemistry C, 2017, 121(3): 1964–1973. doi: 10.1021/acs.jpcc.6b11337 -

 下载:
下载:





 点击查看大图
点击查看大图
计量
- 文章访问数: 9529
- HTML全文浏览量: 3054
- PDF下载量: 85
