




Fabrication of Submicron Tetragonal Polycrystalline ZrO2 by the Transformation of Micro Monoclinic ZrO2 under High Pressure
-
摘要: 高压相变已逐渐发展成为一种制备纳米/亚微米多晶陶瓷块体材料的有效方法。高压可以抑制原子的长程扩散进而抑制晶粒长大,高压下截获的新相不受初始材料晶粒尺寸的制约,通过热力学调控可以得到晶粒尺寸更小的多晶块体材料。陶瓷材料在特定热力学条件下通常会发生相变,新相的形成要经历形核、生长的过程。采用晶粒尺寸为2
$ {\text{μ}} $ m的单斜ZrO2与晶粒尺寸为50 nm的Y2O3以97 : 3的摩尔比混合,在5.5 GPa、800~1700 ℃温压区间内对初始材料进行烧结,采用X射线衍射、扫描电镜、透射电镜对所得样品进行表征。研究结果表明:高压下截获了单斜相和亚微米四方相复合的多晶ZrO2块体材料,1200、1400、1600和1700 ℃温度下获得的四方相的平均晶粒尺寸为(145±62) nm、(246±165) nm、(183±62) nm和(245±107) nm。利用高压相变以微米晶制备细晶粒多晶块体材料,可以避免常规方法中以纳米粉末为初始材料制备细晶粒多晶块体材料存在的团聚、吸附及晶粒长大的问题,进而发展一种以微米晶为初始材料通过高压相变制备高性能细晶粒多晶块体材料的方法。Abstract: The transformation-assisted consolidation under pressure has been demonstrated to be a promising method to fabricate the nano or submicron polycrystalline ceramic materials. The high pressure suppresses the long-range diffusion of the atoms and, consequently, restrains the grain coarsening. The new phases produced at high pressure could show finer grains under the appropriate thermodynamic conditions, which are not subject to the grain size of the raw materials. Ceramic materials exhibit the existence of the transformations under certain thermodynamic conditions and the formation of new phases generally undergoes the nucleation and growth. In the present work, monoclinic microcrystal ZrO2 with average grain size of 2 µm and Y2O3 with average grain size of 50 nm were mixed in molar ratio of 97∶3. The preparation of the samples was carried out by sintering at 5.5 GPa and temperatures of 800–1700 °C using the high pressure cubic cell, and the sample characterization was performed via the X-ray diffraction, scanning electron microscope and transmission electron microscopy. It was found that the monoclinic and submicron tetragonal composite polycrystalline ZrO2 in bulk is obtained under high pressure and high temperature. The average grain size of tetragonal ZrO2 fabricated at 1200, 1400, 1600 and 1700 °C is (145±62) nm, (246±165) nm, (183±62) nm and (245±107) nm, respectively. The synthesis of the fine-grained polycrystalline materials by the transformation under high pressure can solve the problems of agglomeration, adsorption and grain coarsening caused by the nanopowders as the starting materials in the conventional approach, which would be an alternative route to fabricate the fine-grained polycrystalline materials with the enhanced performances.-
Key words:
- high pressure /
- phase transformation /
- micro crystal /
- submicron crystal /
- ZrO2
-
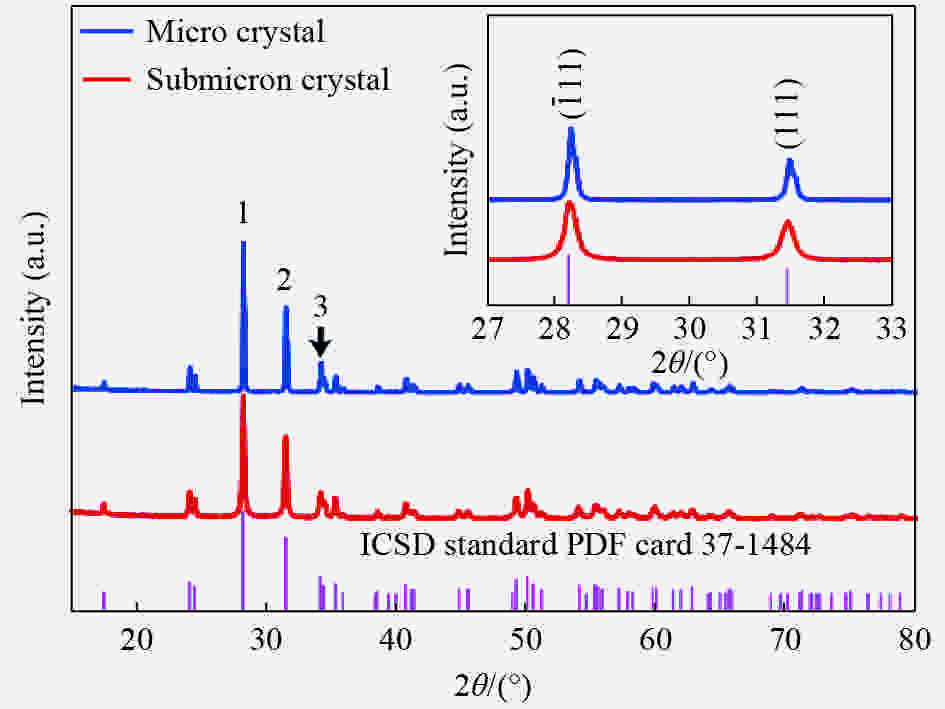
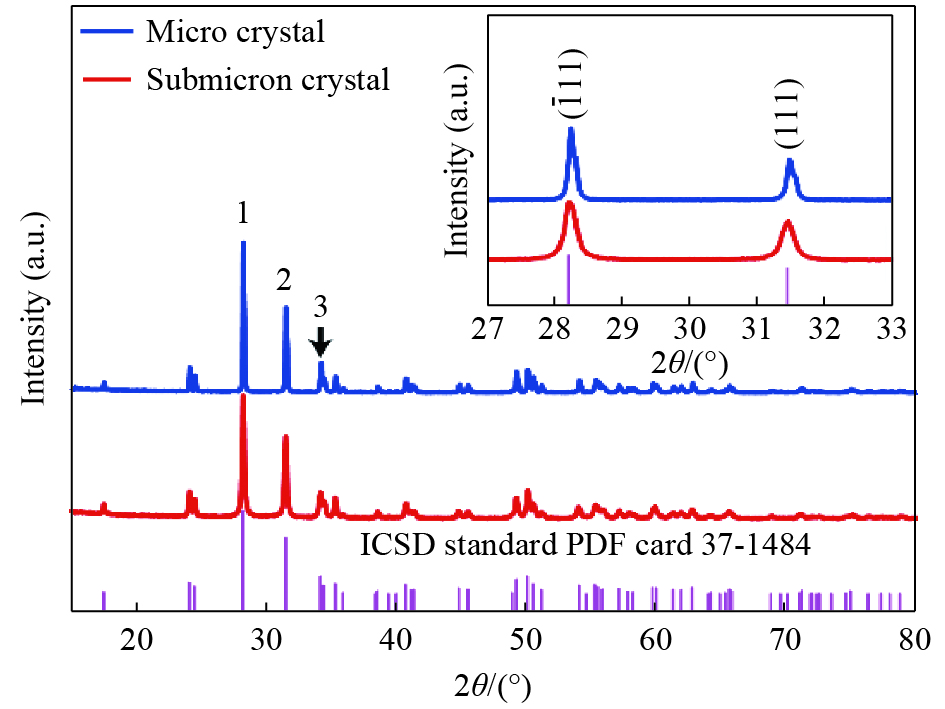
图 2 亚微米原料和微晶ZrO2的XRD图谱(插图为ZrO2单斜相特征峰XRD图谱)
Figure 2. XRD pattern of submicron and microcrystalline ZrO2 (Inset: XRD pattern of the characteristic diffraction peaks of monoclinic ZrO2)

图 3 亚微米晶ZrO2(a)和微米晶ZrO2(b)的SEM照片
Figure 3. SEM images of submicron (a) and microcrystalline ZrO2 (b)
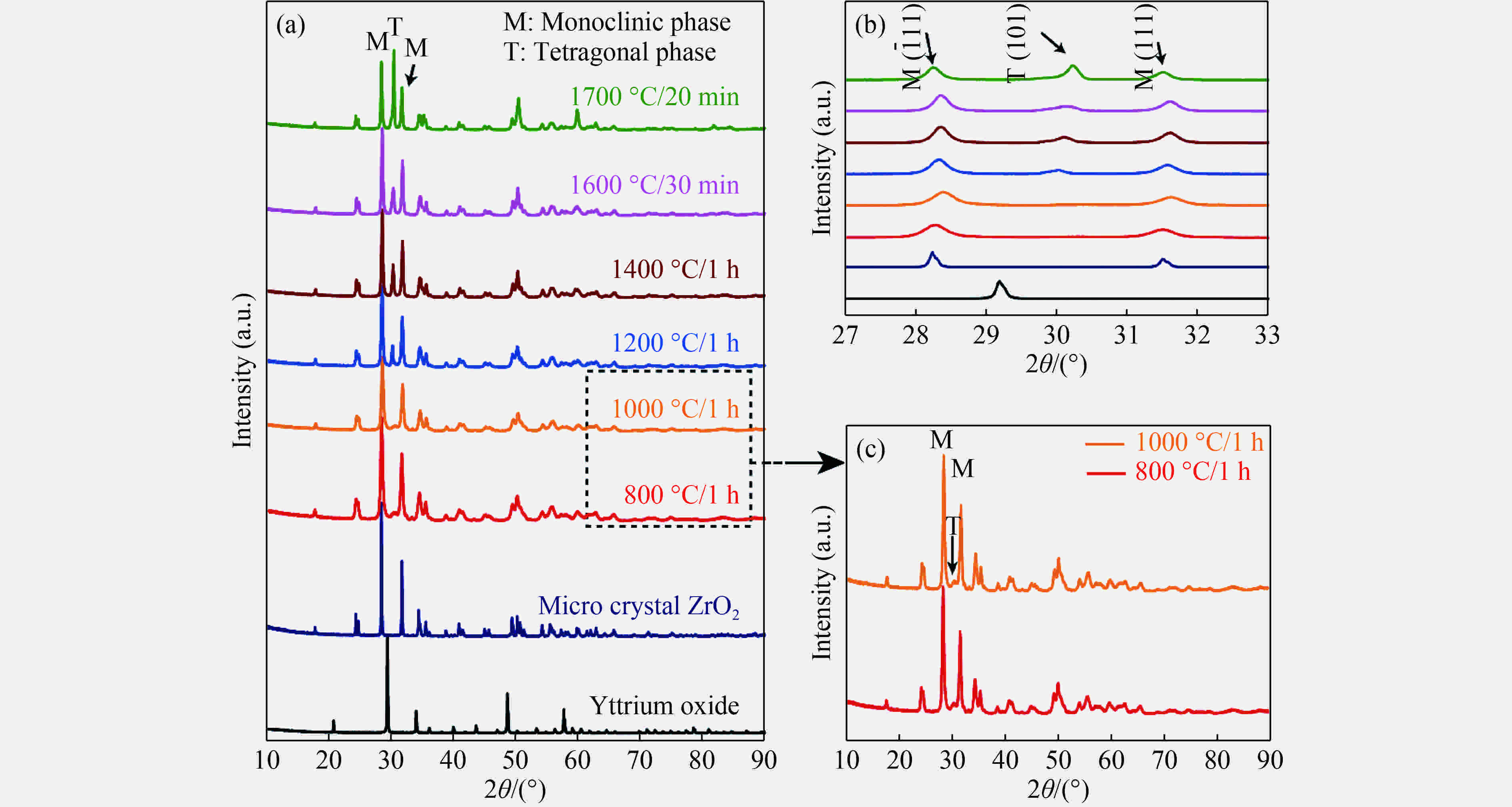
图 4 5.5 GPa不同温度条件下样品的XRD图谱(a)、单斜相和四方相的特征峰放大图谱(b)以及(a)中方框标记的800和1000 ℃条件下合成样品的XRD图谱(c)
Figure 4. (a) XRD pattern of samples sintered at 5.5 GPa and different temperatures; (b) magnified range of the characteristic diffraction peaks of monoclinic and tetragonal phases from (a); (c) XRD patterns of samples sintered at 800 and 1000 ℃ marked with dotted line in (a)

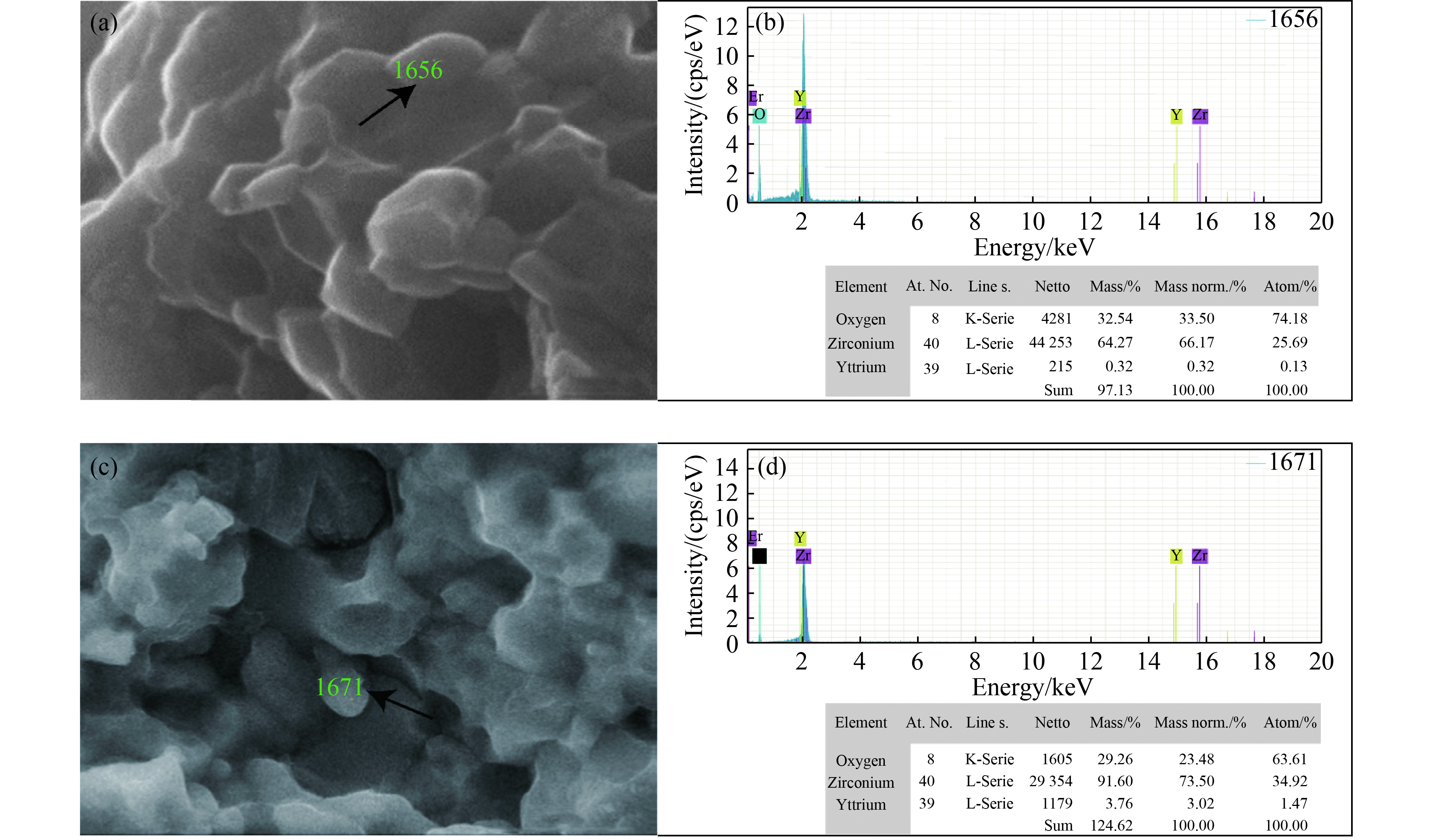
图 5 (a) 1400 ℃温度条件下样品中亚微米晶粒的SEM照片,(b) (a)中箭头所指亚微米晶粒的EDS能谱,(c) 1700 ℃温度条件下样品中亚微米晶粒的SEM照片,(d) (c)中箭头所指亚微米晶粒的EDS能谱
Figure 5. (a) SEM images of submicron grains in the sample sintered at 1400 ℃; (b) EDS spectra of the submicron grain directed by the arrow; (c) SEM images of submicron grains in the sample sintered at 1700 ℃; (d) EDS spectra of the submicron grain directed by the arrow

图 6 5.5 GPa、不同温度下合成样品的SEM照片
Figure 6. SEM images of samples sintered at the condition of 5.5 GPa and different temperatures
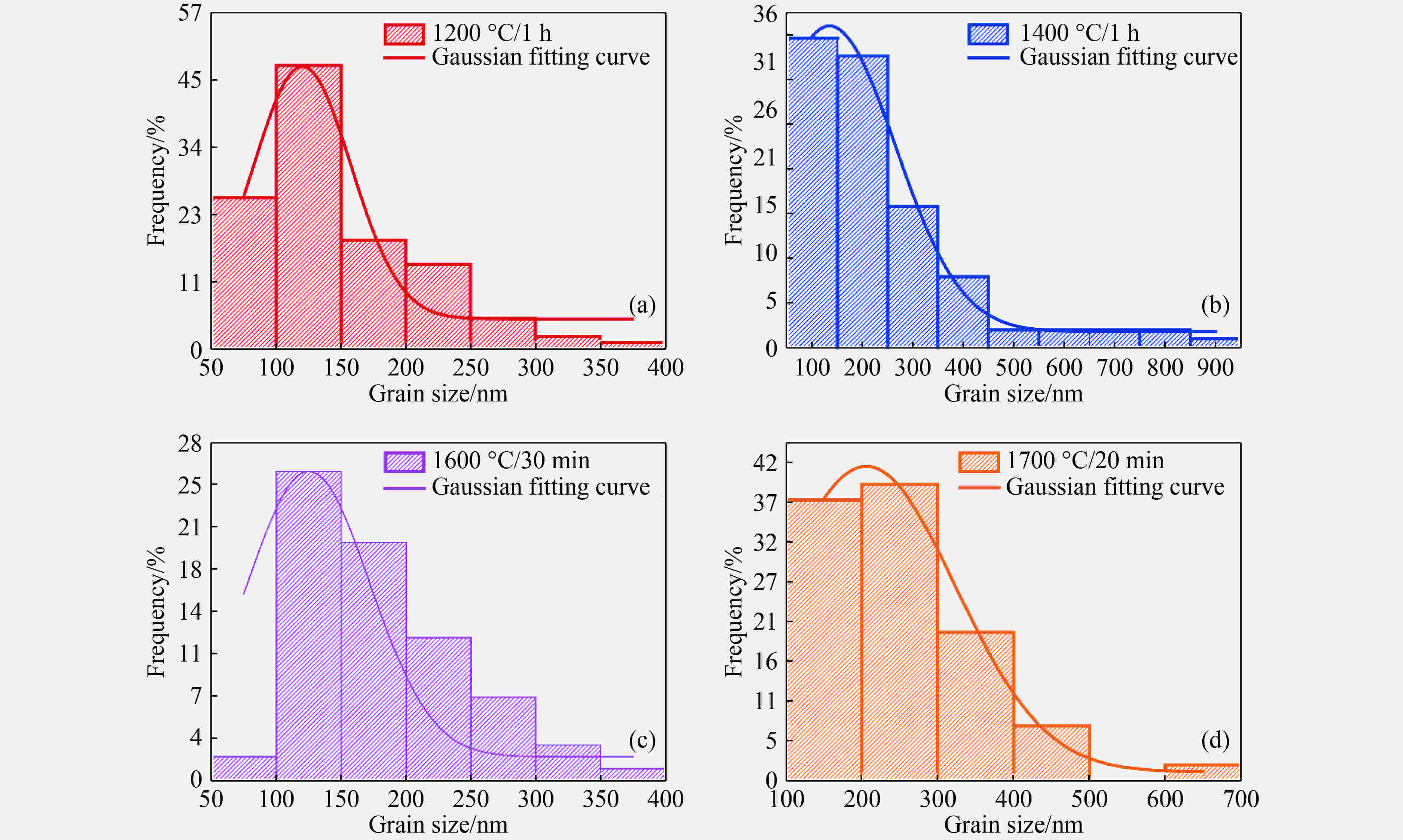
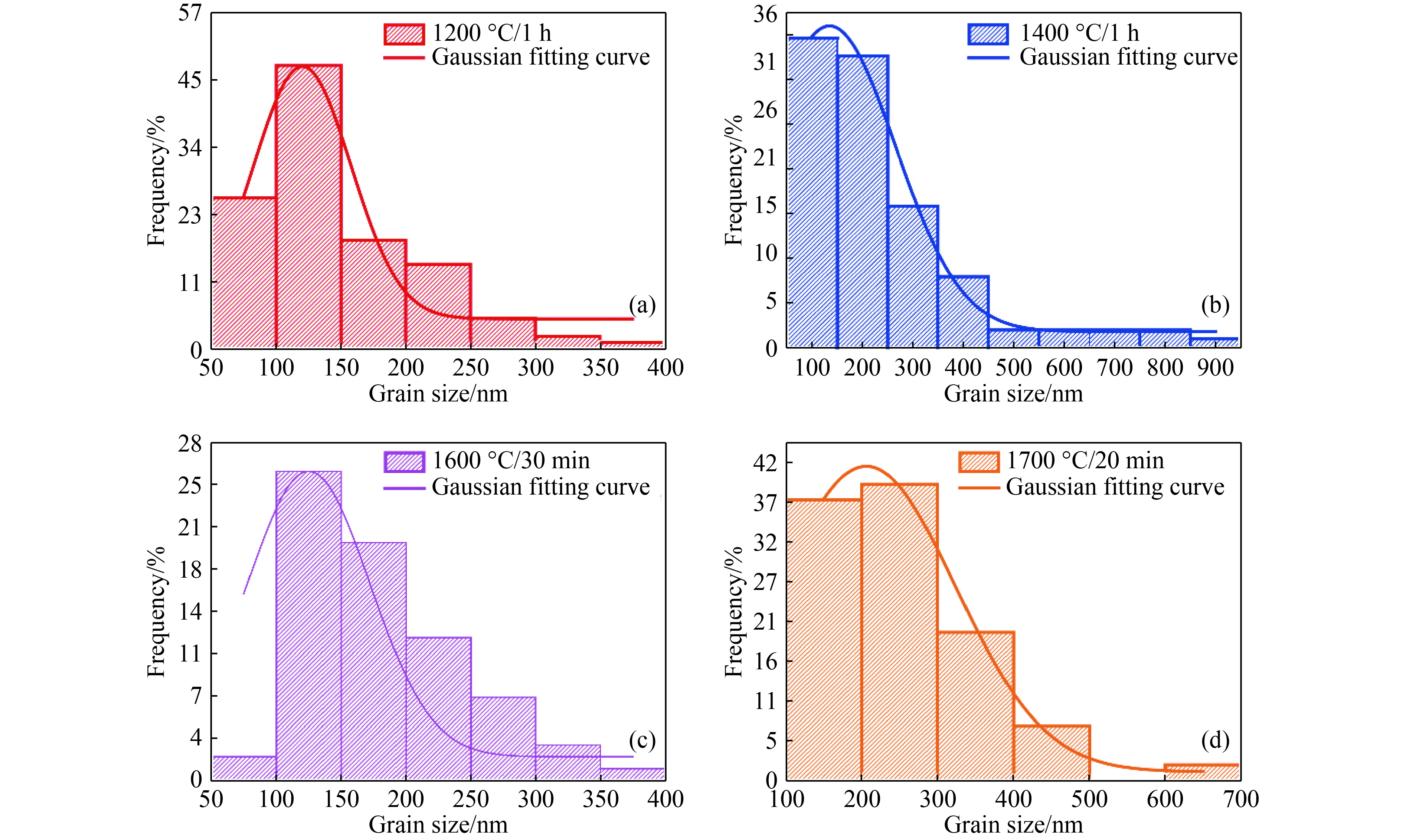
图 7 5.5 GPa不同温度下合成样品中亚微米晶粒的晶粒尺寸分布
Figure 7. Grain size distribution of submicron grains in samples sintered at 5.5 GPa and different temperatures

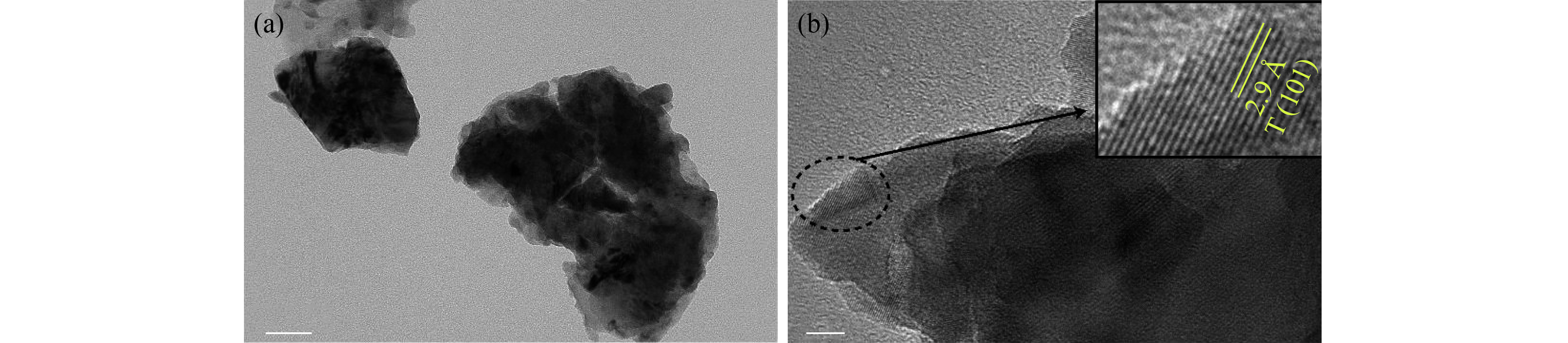
图 8 5.5 GPa、1700 ℃合成样品的TEM照片((a)样品中亚微米晶粒的低倍TEM照片;(b)样品中亚微米晶粒的HRTEM照片,插图为虚线椭圆部位的放大图)
Figure 8. TEM images of the sample sintered at 5.5 GPa and 1700 ℃ ((a) Low-magnification TEM image of submicron grains in the sample; (b) HRTEM image of submicron grains in the sample. Inset: Magnified HRTEM image marked with the dotted ellipse in (b))
表 1 室温下稳定四方相氧化锆的晶胞参数
Table 1. Unit-cell parameters of tetragonal zirconia stabilized at room temperature
 下载: 导出CSV
下载: 导出CSV
-
[1] HALL E O. The deformation and ageing of mild steel: III discussion of results [J]. Proceedings of the Physical Society Section B, 1951, 64(9): 747–753. doi: 10.1088/0370-1301/64/9/303 [2] PETCH N J. The cleavage strength of polycrystals [J]. Journal of the Iron and Steel Institute, 1953, 174: 25–28. [3] SOLOZHENKO V L, KURAKEVYCH O O, LE GODEC Y. Creation of nanostuctures by extreme conditions: high-pressure synthesis of ultrahard nanocrystalline cubic boron nitride [J]. Advanced Materials, 2012, 24(12): 1540–1544. doi: 10.1002/adma.201104361 [4] LIU G, KOU Z, YAN X, et al. Submicron cubic boron nitride as hard as diamond [J]. Applied Physics Letters, 2015, 106(12): 121901. doi: 10.1063/1.4915253 [5] TIAN Y, XU B, YU D, et al. Ultrahard nanotwinned cubic boron nitride [J]. Nature, 2013, 493(7432): 385–388. doi: 10.1038/nature11728 [6] 徐波, 田永君. 纳米孪晶超硬材料的高压合成 [J]. 物理学报, 2017, 66(3): 036201XU B, TIAN Y J. High pressure synthesis of nanotwinned ultrahard materials [J]. Acta Physica Sinica, 2017, 66(3): 036201 [7] IRIFUNE T, KURIO A, SAKAMOTO S, et al. Ultrahard polycrystalline diamond from graphite [J]. Nature, 2003, 421(6923): 599–600. [8] 王海阔, 张相法, 位星, 等. 直接转化法合成大尺寸纯相多晶金刚石 [J]. 金刚石与磨料磨具工程, 2018, 38(1): 1–6WANG H K, ZHANG X F, WEI X, et al. Synthesizing bulk polycrystalline diamond by method of direct phase transition [J]. Diamond & Abrasives Engineering, 2018, 38(1): 1–6 [9] HUANG Q, YU D, XU B, et al. Nanotwinned diamond with unprecedented hardness and stability [J]. Nature, 2014, 510(7504): 250. doi: 10.1038/nature13381 [10] EHRE D, GUTMANAS E Y, CHAIM R. Densification of nanocrystalline MgO ceramics by hot-pressing [J]. Journal of the European Ceramic Society, 2005, 25(16): 3579–3585. doi: 10.1016/j.jeurceramsoc.2004.09.023 [11] TANG F, HAGIWARA M, SCHOENUNG J M. Formation of coarse-grained inter-particle regions during hot isostatic pressing of nanocrystalline powder [J]. Scripta Materialia, 2005, 53(6): 619–624. doi: 10.1016/j.scriptamat.2005.05.034 [12] BINNER J, ANNAPOORANI K, PAUL A, et al. Dense nanostructured zirconia by two stage conventional/hybrid microwave sintering [J]. Journal of the European Ceramic Society, 2008, 28(5): 973–977. doi: 10.1016/j.jeurceramsoc.2007.09.002 [13] DAHL P, KAUS I, ZHAO Z, et al. Densification and properties of zirconia prepared by three different sintering techniques [J]. Ceramics International, 2007, 33(8): 1603–1610. doi: 10.1016/j.ceramint.2006.07.005 [14] SALAMON D, KALOUSEK R, MACA K, et al. Rapid grain growth in 3Y-TZP nanoceramics by pressure-assisted and pressure-less SPS [J]. Journal of the American Ceramic Society, 2015, 98(12): 3706–3712. doi: 10.1111/jace.13837 [15] MAZAHERI M, ZAHEDI A M, HEJAZI M M. Processing of nanocrystalline 8 mol% yttria-stabilized zirconia by conventional, microwave-assisted and two-step sintering [J]. Materials Science and Engineering A, 2008, 492(1/2): 261–267. [16] ZHANG L, WANG Y, LV J, et al. Erratum: materials discovery at high pressures [J]. Nature Reviews Materials, 2017, 2(4): 17005. doi: 10.1038/natrevmats.2017.5 [17] YAVETSKIY R P, BAUMER V N, DANYLENKO M I, et al. Transformation-assisted consolidation of Y2O3: Eu3+ nanospheres as a concept to optical nanograined ceramics [J]. Ceramics International, 2014, 40(2): 3561–3569. doi: 10.1016/j.ceramint.2013.09.072 [18] WOLLMERSHAUSER J A, FEIGELSON B N, GORZKOWSKI E P, et al. An extended hardness limit in bulk nanoceramics [J]. Acta Materialia, 2014, 69(5): 9–16. [19] KEAR B H, COLAIZZI J, MAYO W E, et al. On the processing of nanocrystalline and nanocomposite ceramics [J]. Scripta Materialia, 2001, 44(8/9): 2065–2068. [20] LIAO S C, COLAIZZI J, CHEN Y, et al. Refinement of nanoscale grain structure in bulk titania via a transformation-assisted consolidation (TAC) method [J]. Journal of the American Ceramic Society, 2000, 83(9): 2163–2169. [21] LIAO S C, CHEN Y J, MAYO W E, et al. Transformation-assisted consolidation of bulk nanocrystalline TiO2 [J]. Nanostructured Materials, 1999, 11(4): 553–557. doi: 10.1016/S0965-9773(99)00344-X [22] LIAO S C, PAE K D, MAYO W E. Retention of nanoscale grain size in bulk sintered materials via a pressure-induced phase transformation [J]. Nanostructured Materials, 1997, 8(6): 645–656. doi: 10.1016/S0965-9773(97)00227-4 [23] LIAO S C, PAE K D, MAYO W E. High pressure and low temperature sintering of bulk nanocrystalline TiO2 [J]. Materials Science and Engineering A, 1995, 204(1/2): 152–159. [24] DENRY I, KELLY J R. State of the art of zirconia for dental applications [J]. Dental Materials, 2008, 24(3): 299–307. doi: 10.1016/j.dental.2007.05.007 [25] RÜHLE M. Microscopy of structural ceramics [J]. Advanced Materials, 1997, 9(3): 195–217. doi: 10.1002/adma.19970090304 [26] YASHIMA M, HIROSE T, KAKIHANA M, et al. Size and charge effects of dopant M on the unit-cell parameters of monoclinic zirconia solid solutions Zr0.98M0.02O2-δ (M= Ce, La, Nd, Sm, Y, Er, Yb, Sc, Mg, Ca) [J]. Journal of the American Ceramic Society, 1997, 80(1): 171–175. doi: 10.1111/j.1151-2916.1997.tb02806.x [27] DENRY I, KELLY J R. Emerging ceramic-based materials for dentistry [J]. Journal of Dental Research, 2014, 93(12): 1235–1242. doi: 10.1177/0022034514553627 [28] PORTER D L, HEUER A H. Mechanisms of toughening partially stabilized zirconia (PSZ) [J]. Journal of the American Ceramic Society, 1977, 60(3/4): 183–184. [29] WHITNEY E D. Effect of pressure on monoclinic-tetragonal transition of zirconia: thermodynamics [J]. Journal of the American Ceramic Society, 1962, 45(12): 612–613. doi: 10.1111/jace.1962.45.issue-12 [30] WHITNEY E D. Electrical resistivity and diffusionless phase transformations of zirconia at high temperatures and ultrahigh pressures [J]. Journal of the Electrochemical Society, 1965, 112(1): 91–94. doi: 10.1149/1.2423476 [31] VAHLDIEK F W, ROBINSON L B, LYNCH C T. Tetragonal zirconium oxide prepared under high pressure [J]. Science, 1963, 142(3595): 1059–1060. doi: 10.1126/science.142.3595.1059 [32] KULCINSKI G L. High-pressure induced phase transition in ZrO2 [J]. Journal of the American Ceramic Society, 1968, 51(10): 582–583. doi: 10.1111/jace.1968.51.issue-10 [33] ALZYAB B, PERRY C H, INGEL R P. High-pressure phase transitions in zirconia and yttria-doped zirconia [J]. Journal of the American Ceramic Society, 1987, 70(10): 760–765. doi: 10.1111/jace.1987.70.issue-10 [34] RHODES W H. Agglomerate and particle size effects on sintering yttria-stabilized zirconia [J]. Journal of the American Ceramic Society, 1981, 64(1): 19–22. doi: 10.1111/jace.1981.64.issue-1 [35] MAGLIA F, TREDICI I G, ANSELMI-TAMBURINI U. Densification and properties of bulk nanocrystalline functional ceramics with grain size below 50 nm [J]. Journal of the European Ceramic Society, 2013, 33(6): 1045–1066. doi: 10.1016/j.jeurceramsoc.2012.12.004 [36] 王海阔, 任瑛, 贺端威, 等. 六面顶压机立方压腔内压强的定量测量及受力分析 [J]. 物理学报, 2017, 66(9): 090702WANG H K, REN Y, HE D W, et al. Force analysis and pressure quantitative measurement for the high pressure cubic cell [J]. Acta Physica Sinica, 2017, 66(9): 090702 [37] ANDERSSON G, SUNDQVIST B, BÄCKSTRÖM G. A high-pressure cell for electrical resistance measurements at hydrostatic pressures up to 8 GPa: results for Bi, Ba, Ni, and Si [J]. Journal of Applied Physics, 1989, 65(10): 3943–3950. doi: 10.1063/1.343360 [38] 王海阔, 贺端威, 许超, 等. 基于国产铰链式六面顶压机的大腔体静高压技术研究进展 [J]. 高压物理学报, 2013, 27(5): 633–661WANG H K, HE D W, XU C, et al. Development of large volume-high static pressure techniques based on the hinge-type cubic presses [J]. Chinese Journal of High Pressure Physics, 2013, 27(5): 633–661 [39] 陈晓芳, 贺端威, 王福龙, 等. 基于铰链式六面顶压机的二级6-8模超高压大腔体内置加热元件的设计与温度标定 [J]. 高压物理学报, 2009, 23(2): 98–104 doi: 10.3969/j.issn.1000-5773.2009.02.004CHEN X F, HE D W, WANG F L, et al. Design and temperature calibration for heater cell of split-sphere high pressure apparatus based on the hinge-type cubic-anvil Press [J]. Chinese Journal of High Pressure Physics, 2009, 23(2): 98–104 doi: 10.3969/j.issn.1000-5773.2009.02.004 [40] TORAYA H, YOSHIMURA M, SOMIYA S. Calibration curve for quantitative analysis of the monoclinic-tetragonal ZrO2 system by X-ray diffraction [J]. Journal of the American Ceramic Society, 1984, 67(6): C-119–C-121. [41] KROGSTAD J A, LEPPLE M, GAO Y, et al. Effect of yttria content on the zirconia unit cell parameters [J]. Journal of the American Ceramic Society, 2011, 94(12): 4548–4555. doi: 10.1111/j.1551-2916.2011.04862.x [42] IGAWA N, ISHII Y, NAGASAKI T, et al. Crystal structure of metastable tetragonal zirconia by neutron powder diffraction study [J]. Journal of the American Ceramic Society, 1993, 76(10): 2673–2676. doi: 10.1111/jace.1993.76.issue-10 [43] MICHEL D, MAZEROLLES L, JORBA M P Y. Fracture of metastable tetragonal zirconia crystals [J]. Journal of Materials Science, 1983, 18(9): 2618–2628. doi: 10.1007/BF00547578 -


 下载:
下载:




 点击查看大图
点击查看大图
计量
- 文章访问数: 7835
- HTML全文浏览量: 4298
- PDF下载量: 42
