




First-Principles Investigations on Metallic Silicon Allotropes
-
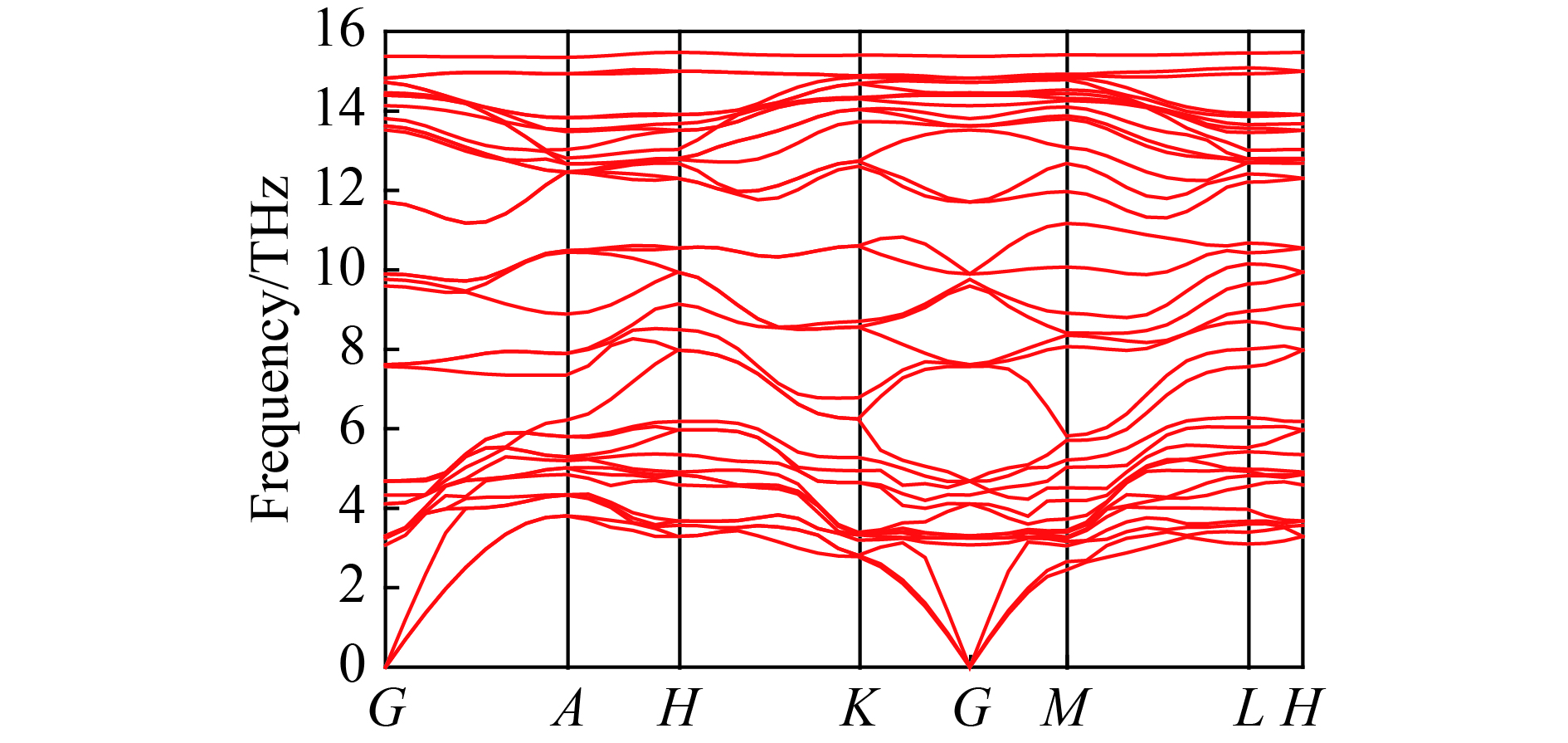
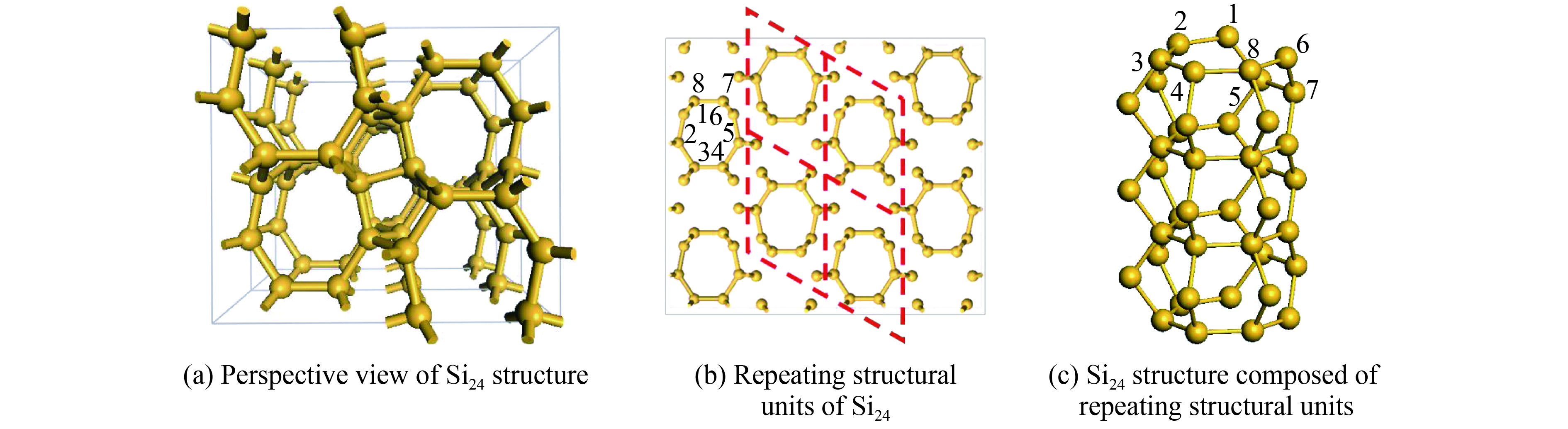
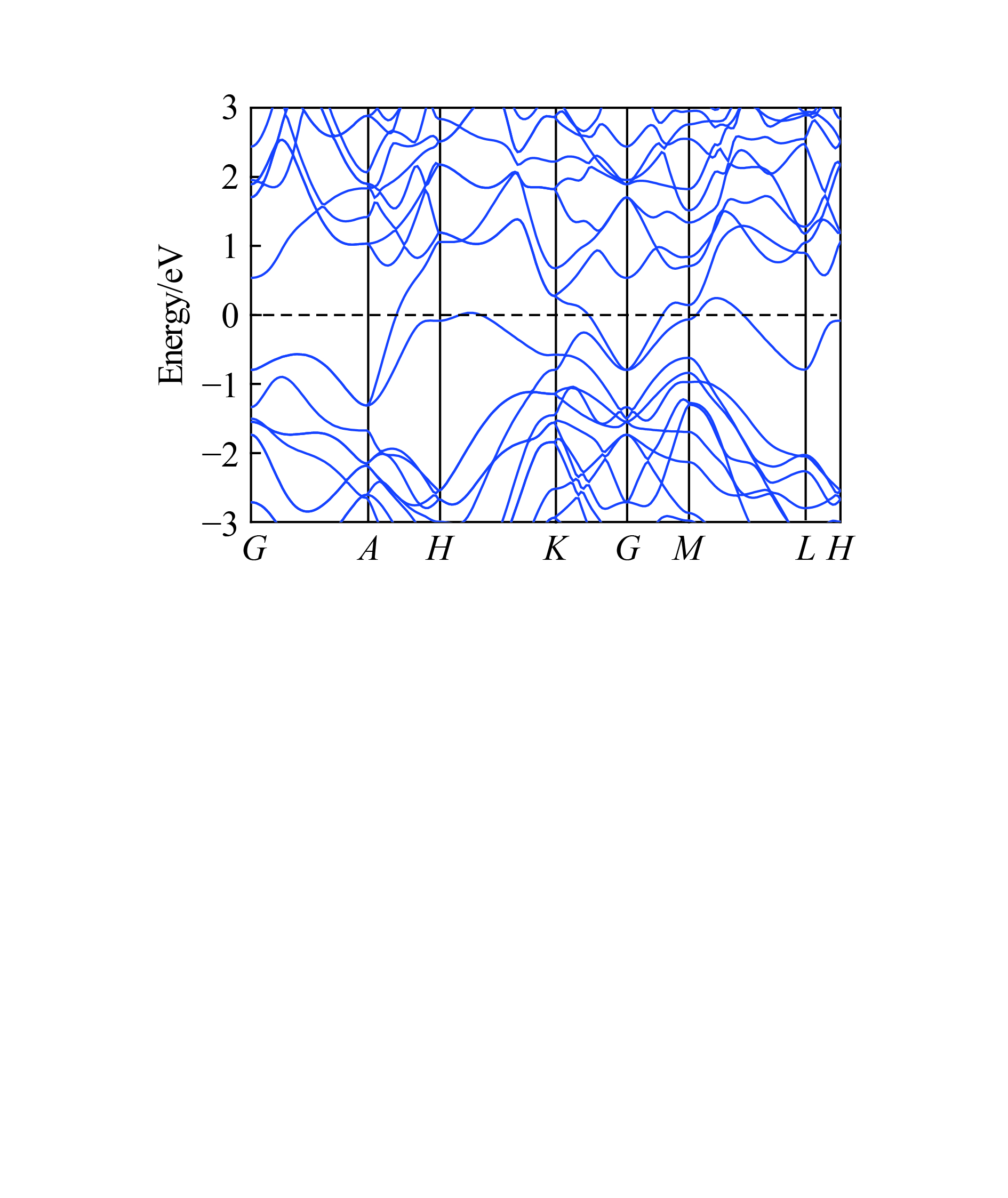
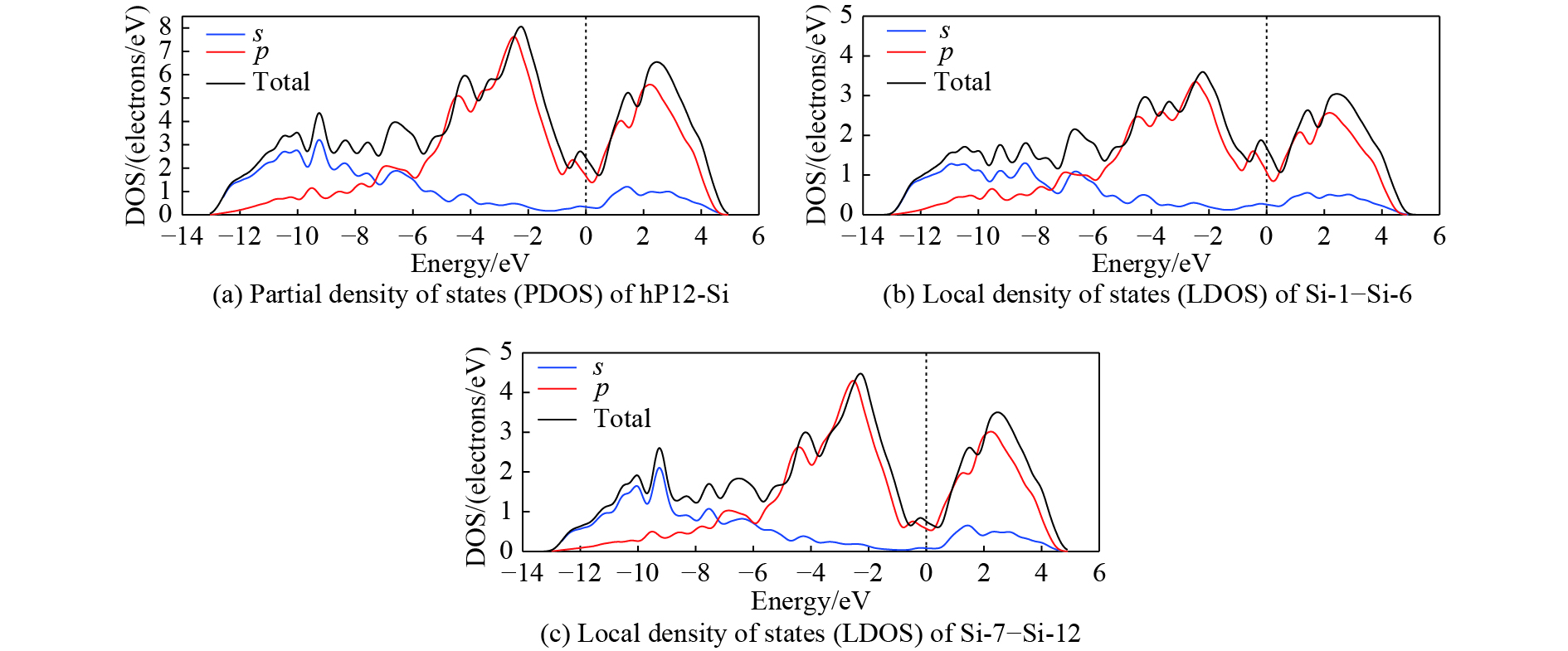
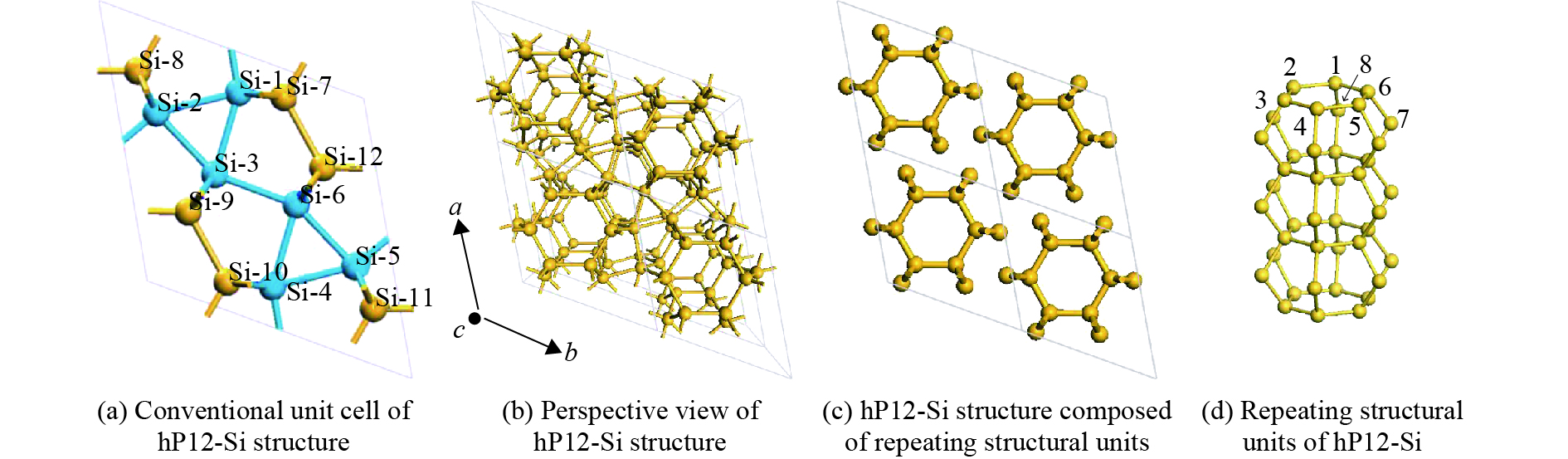
摘要: 从理论上提出了一种新型金属性硅的同素异形体hP12-Si。 hP12-Si结构可以看作是由六元环形成的一种隧道型结构,与之前报道的Si24结构近似。弹性常数和声子谱的计算结果验证了该结构在常压下的稳定性。通过结构遗传性和热力学稳定性分析表明,可以效仿Si24的制备方法,通过预先合成出高压前驱物LiSi12再除去其中的Li原子来获得hP12-Si。在这种结构中, 有一半的硅原子为5配位,其他硅原子为4配位。电子结构计算表明,该结构具有金属导电性,导电性主要是由于5配位原子的存在导致价电子具有离域性。Abstract: A new metallic metastable silicon allotrope hP12-Si has been theoretically proposed using the particle swarm optimization method. The hP12-Si structure can be seen as a combination of a tunnel-type structure formed from six-membered sp3 silicon rings, which is similar to the previously reported Si24 structure. Its stability was verified by calculating its elastic constants and phonon spectrum. The analysis of structural heritability and thermodynamic stability shows that hP12-Si might be obtained by removing Li atoms from the pre-synthetic LiSi12 precursor, which is analogous with the recent preparation of Si24. There are 50% five coordinated silicon atoms, whereas the others are four coordinated in the hP12-Si structure. Electronic band structure calculation indicated that this structure could perform the metallic properties, which might be resulted from the delocalization of valence electrons caused by the existence of five coordinated atoms.
-
Key words:
- metallic silicon /
- first-principles /
- high density silicon /
- structural transition
-
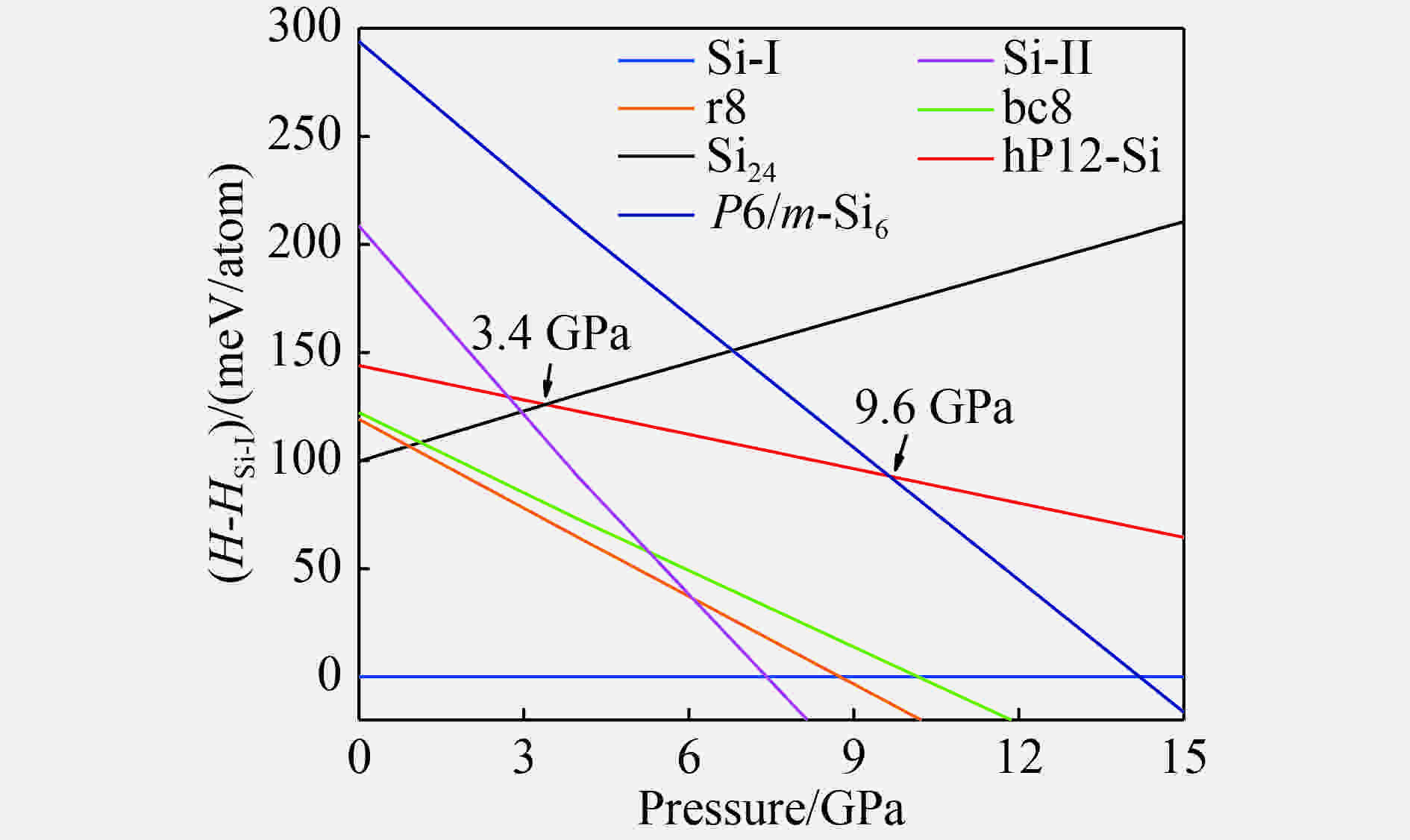
图 3 硅同素异形体相对于Si-I相的焓压曲线
Figure 3. Enthalpies of various Si allotropes relative to Si-I as a function of pressure
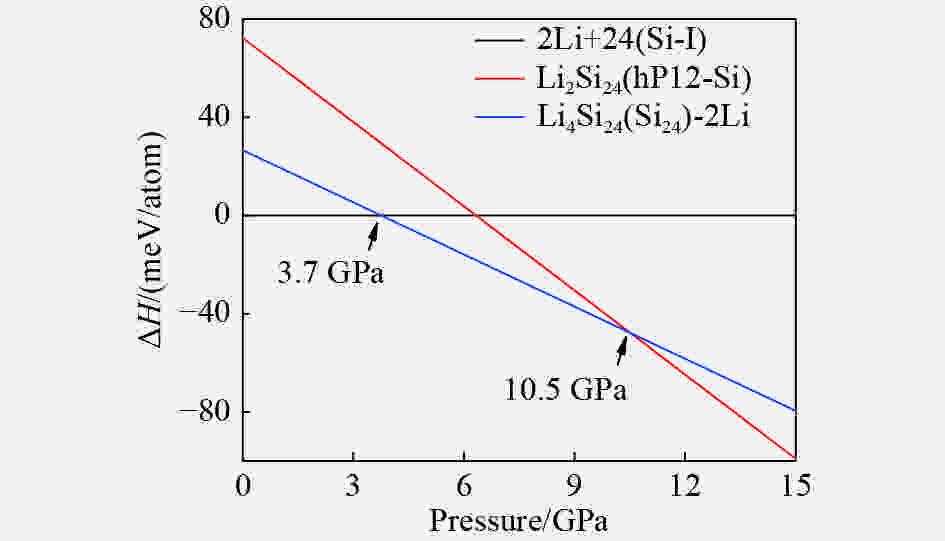
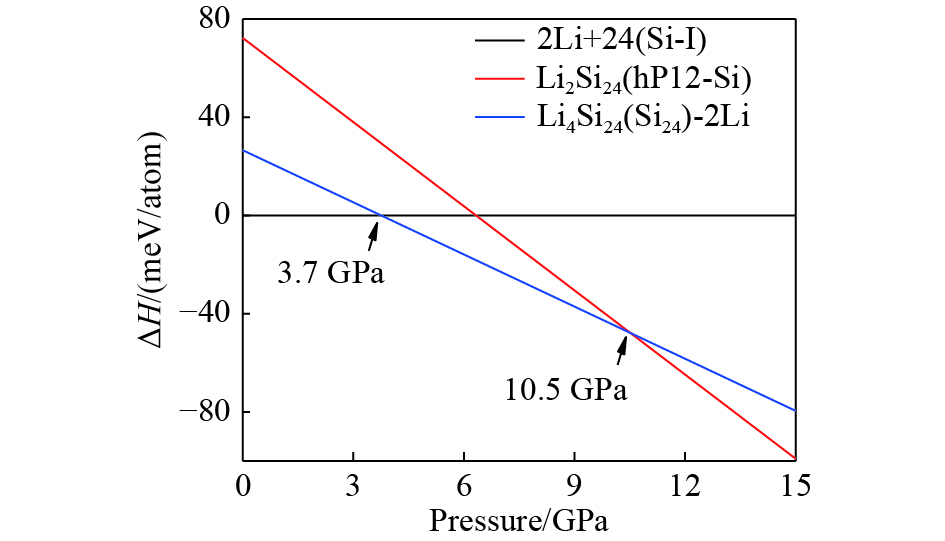
图 5 Li2Si24和Li4Si24原料的焓压曲线
Figure 5. Enthalpies of Li2Si24 and Li4Si24 phase relative to reactants (Li and Si-I) as a function of pressure
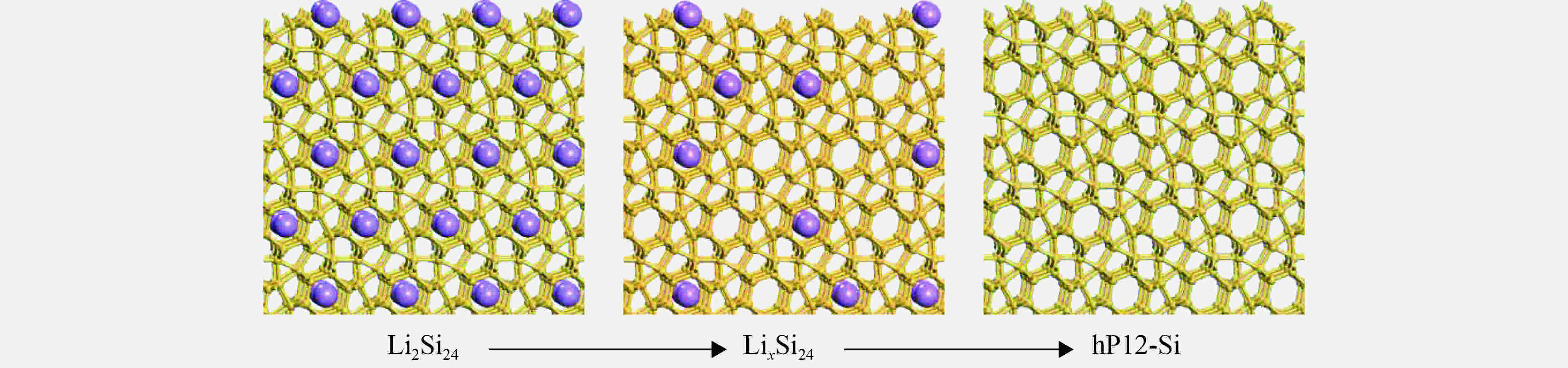
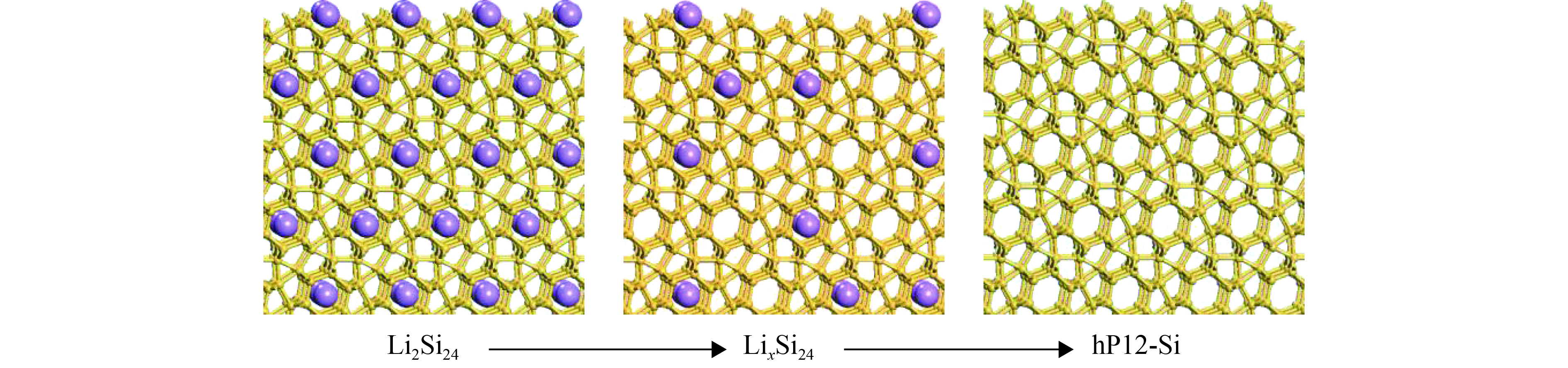
图 6 Li2Si24脱Li过程
Figure 6. Schematic of the compositional change from Li2Si24 (left) to hP12-Si (right)
表 1 常压下hP12-Si单胞的晶体结构数据
Table 1. Crystallographic data for hP12-Si conventional cell
Space group a/Å c/Å ρ/(g·cm–3) Atomic positions P6/m (175) 8.202 3.823 2.512 Si:6k (0.499 33, 0.846 42, 0.5) Si:6j (0.679 02, 0.892 27, 0.0)  下载: 导出CSV
下载: 导出CSV
表 2 hP12-Si单胞中化学键的布居数和键长
Table 2. Populations and length of silicon bond in hP12-Si conventional cell
Silicon bond Populations Length of silicon bond/Å Si-7—Si-11, Si-7—Si-12, Si-8—Si-10 0.76 2.321 Si-8—Si-12, Si-9—Si-10, Si-9—Si-11 Si-1—Si-7, Si-2—Si-8, Si-3—Si-9 0.77 2.327 Si-4—Si-10, Si-5—Si-11, Si-6—Si-12 Si-1—Si-2, Si-1—Si-3, Si-2—Si-3 0.33 2.462 Si-4—Si-5, Si-4—Si-6, Si-5—Si-6 Si-1—Si-4, Si-2—Si-5, Si-3—Si-6 0.41 2.514
下载: 导出CSV
表 3 常压下hP12-Si、Si24和Si-I结构的密度、弹性常数、体弹性模量和剪切模量
Table 3. Calculated density, elastic constants , bulk moduli, and shear moduli of hP12-Si, Si24 and Si-I at ambient pressure
Structure ρ/(g·cm–3) Elastic constants/GPa B/GPa G/GPa C11 C22 C33 C44 C55 C66 C12 C13 C23 hP12-Si 2.512 166 166 148 51 51 56 66 66 94 94 51 Si24 2.236 164 204 147 37 42 51 40 46 85 85 50 Si-I 2.402 161 161 161 76 76 76 62 62 95 95 64
下载: 导出CSV
表 4 常压下hP12-Si结构的原子位置、配位数和布居数
Table 4. Wyckoff positions, coordination numbers, and populations of silicon atoms in hP12-Si
Atomic number Wyckoff positions Coordination numbers Populations Si-1–Si-6 6k (0.499 33, 0.846 42, 0.5) 5 –0.02 Si-7–Si-12 6j (0.679 02, 0.892 27, 0.0) 4 0.02
下载: 导出CSV
-
[1] KILBY J S. Miniaturized electronic circuits [J]. Google Patents, 1964. [2] NOYCE R N. Semiconductor device-and-lead structure: US 2981877 [P]. 1961–04–25. [3] CARLSON D E, WRONSKI C R. Amorphous silicon solar cell [J]. Applied Physics Letters, 1976, 28(11): 671–673. doi: 10.1063/1.88617 [4] GREEN M A. Solar cells: operating principles, technology, and system applications [J]. Prentice-Hall, 1982. [5] GREEN M A, EMERY K, HISHIKAWA Y, et al. Solar cell efficiency tables (Version 45) [J]. Progress in Photovoltaics: Research and Applications, 2015, 23(1): 1–9. doi: 10.1002/pip.v23.1 [6] MUJICA A, RUBIO A, MUNOZ A, et al. High-pressure phases of group-IV, III-V, and H-VI compounds [J]. Reviews of Modern Physics, 2003, 75(3): 863–912. doi: 10.1103/RevModPhys.75.863 [7] BAUTISTA-HERNANDEZ A, RANGEL T, ROMERO A H, et al. Structural and vibrational stability of M and Z phases of silicon and germanium from first principles [J]. Journal of Applied Physics, 2013, 113(19): 193504. doi: 10.1063/1.4804668 [8] FUJIMOTO Y, KORETSUNE T, SAITO S, et al. A new crystalline phase of four-fold coordinated silicon and germanium [J]. New Journal of Physics, 2008, 10(8): 083001. [9] WU F, JUN D, KAN E, et al. Density functional predictions of new silicon allotropes: electronic properties and potential applications to Li-battery anode materials [J]. Solid State Communications, 2011, 151(18): 1228–1230. doi: 10.1016/j.ssc.2011.06.001 [10] ZHAO Z, TIAN F, DONG X, et al. Tetragonal allotrope of group 14 elements [J]. Journal of the American Chemical Society, 2012, 134(30): 12362–12365. doi: 10.1021/ja304380p [11] MALONE B D, SAU J D, COHEN M L. Ab initio survey of the electronic structure of tetrahedrally bonded phases of silicon [J]. Physical Review B, 2008, 78(3): 35210. doi: 10.1103/PhysRevB.78.035210 [12] FAN Q, CHAI C, WEI Q, et al. Novel silicon allotropes: stability, mechanical, and electronic properties [J]. Journal of Applied Physics, 2015, 118(18): 185704. doi: 10.1063/1.4935549 [13] LUO K, ZHAO Z, MA M, et al. Si10: a sp3 silicon allotrope with spirally connected Si5 tetrahedrons [J]. Chemistry of Materials, 2016, 28(18): 6441–6445. doi: 10.1021/acs.chemmater.6b02484 [14] XIANG H J, HUANG B, KAN E, et al. Towards direct-gap silicon phases by the inverse band structure design approach [J]. Physical Review Letters, 2013, 110(11): 13–16. [15] WANG Q, XU B, SUN J, et al. Direct band gap silicon allotropes [J]. Journal of the American Chemical Society, 2014, 136(28): 9826–9829. doi: 10.1021/ja5035792 [16] WANG Q, LUO K, MA M, et al. A new metastable metallic silicon allotrope [J]. Chinese Science Bulletin (Chinese Version), 2015, 60(27): 2616. doi: 10.1360/N972015-00200 [17] WEN Z, LU G, MAO S, et al. Silicon nanotube anode for lithium-ion batteries [J]. Electrochemistry Communications, 2013, 29: 67–70. doi: 10.1016/j.elecom.2013.01.015 [18] SUNG H J, HANG W H, LEE I H, et al. Superconducting open-framework allotrope of silicon at ambient pressure [J]. Physical Review Letters, 2018, 120(15): 157001. doi: 10.1103/PhysRevLett.120.157001 [19] BAI J, ZENG X C, TANAKA H, et al. Metallic single-walled silicon nanotubes [J]. Proceedings of the National Academy of Sciences of the United States of America, 2004, 101(9): 2664–2668. doi: 10.1073/pnas.0308467101 [20] HEVER A, BERNSTEIN J, HOD O. Structural stability and electronic properties of sp3 type silicon nanotubes [J]. The Journal of chemical physics, 2012, 137(21): 214702. doi: 10.1063/1.4767389 [21] LIN C, POVINELLI M L. Optical absorption enhancement in silicon nanowire arrays with a large lattice constant for photovoltaic applications [J]. Optics Express, 2009, 17(22): 19371–19381. doi: 10.1364/OE.17.019371 [22] CLARK S J, SEGALL M D, PICKARD C J, et al. First principles methods using CASTEP [J]. Zeitschrift für Kristallographie–Crystalline Materials, 2005, 220(5/6): 567–570. [23] WANG Y, LV J, ZHU L, et al. CALYPSO: A method for crystal structure prediction [J]. Computer Physics Communications, 2012, 183(10): 2063–2070. doi: 10.1016/j.cpc.2012.05.008 [24] WANG Y, LV J, ZHU L, et al. Crystal structure prediction via particle-swarm optimization [J]. Physical Review B, 2010, 82(9): 094116. [25] LAASONEN K, CAR R, LEE C, et al. Implementation of ultrasoft pseudopotentials in ab initio molecular dynamics [J]. Physical Review B, 1991, 43(8): 6796–6799. doi: 10.1103/PhysRevB.43.6796 [26] VANDERBILT D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism [J]. Physical Review B, 1990, 41(11): 7892–7895. doi: 10.1103/PhysRevB.41.7892 [27] PERDEW J P, ZUNGER A. Self-interaction correction to density-functional approximations for many-electron systems [J]. Physical Review B, 1981, 23(10): 5048–5079. doi: 10.1103/PhysRevB.23.5048 [28] PURVEE B, SADHNA S. Pressure induced structural phase transitions–a review [J]. Central European Journal of Chemistry, 2012, 10(5): 1391–1422. [29] CEPERLEY D M, ALDER B J. Ground state of the electron gas by a stochastic method [J]. Physical Review Letters, 1980, 45(7): 566–569. doi: 10.1103/PhysRevLett.45.566 [30] MONKHORST H J, PACK J D. Special points for Brillouin-zone integrations [J]. Physical Review B, 1976, 13(12): 5188. doi: 10.1103/PhysRevB.13.5188 [31] KRESSE G, FURTHMÜLLER J, HAFNER J. Ab initio force constant approach to phonon dispersion relations of diamond and graphite [J]. Europhysics Letters, 1995, 32(9): 729. doi: 10.1209/0295-5075/32/9/005 [32] TSE J S, KLUG D D, PATCHKOVSKII S, et al. Chemical bonding, electron-phonon coupling, and structural transformations in high-pressure phases of Si [J]. The Journal of Physical Chemistry B, 2006, 110(8): 3721–3726. doi: 10.1021/jp0554341 -

 下载:
下载:


 点击查看大图
点击查看大图
计量
- 文章访问数: 11210
- HTML全文浏览量: 4356
- PDF下载量: 35
