




Evolution of Crystal Structures and Electronic Properties for Ir2P under High Pressure
-
摘要: 在压强为0~100 GPa范围内,运用CALYPSO结构搜索技术,结合基于密度泛函理论中的第一性原理方法,对Ir2P晶体进行结构预测,并对预测出的晶体结构和物理性质进行细致的研究。在常压下,预测得出α-Ir2P相具有立方结构,其空间群为Fm3m,与实验所得结构一致;压强为86.4 GPa时,发生结构相变,由α-Ir2P相转变为β-Ir2P相,为四方结构,其空间群为I4/mmm。在相变过程中,晶体体积发生坍塌,并且出现不连续变化的一级相变。电子性质计算表明,86.4 GPa时,预测的β-Ir2P相中导带和价带在费米面附近发生交叠,表明其结构具有金属性质;电子局域函数计算表明,β-Ir2P相具有丰富的化学键,包括极性共价键、金属键和离子键;Bader电荷转移计算得出,由于Ir原子具有较强的电负性,β-Ir2P相中每个P原子向每个Ir原子电荷转移0.19e。Abstract: The crystals of Ir2P were predicted under the pressure ranging from 0 to 100 GPa using the CALYPSO structure exploration technique with the first-principles method based on the density functional theory. The predicted physical properties and crystal structures were examined in detail. At ambient pressure, the predicted α-Ir2P phase was found to have a cubic structure with Fm3m space group, which is consistent with the experimental structure. The pressure-induced structural transformations were unraveled, from the α-Ir2P phase to the β-Ir2P phase at 86.4 GPa. The predicted β-Ir2P phase has I4/mmm space group. In the process of phase transition, the volume of the crystal collapses and a discontinuous first order phase transition occurred. The calculation of the electronic properties showed that the predicted conduction bands and the valence bands of the β-Ir2P phase overlap near the Fermi surface at 86.4 GPa, indicating that the structure of the β-Ir2P phase has metallic properties. The electron localization function revealed that the β-Ir2P phase has a polar covalent bond, a metallic bond and an ionic bond. The Bader charge transfer calculations showed that each P atom transfers 0.19e to Ir atom, mainly due to the strong electronegativity of the Ir atoms.
-
Key words:
- high pressure /
- first-principles /
- crystal structure prediction /
- Ir2P
-
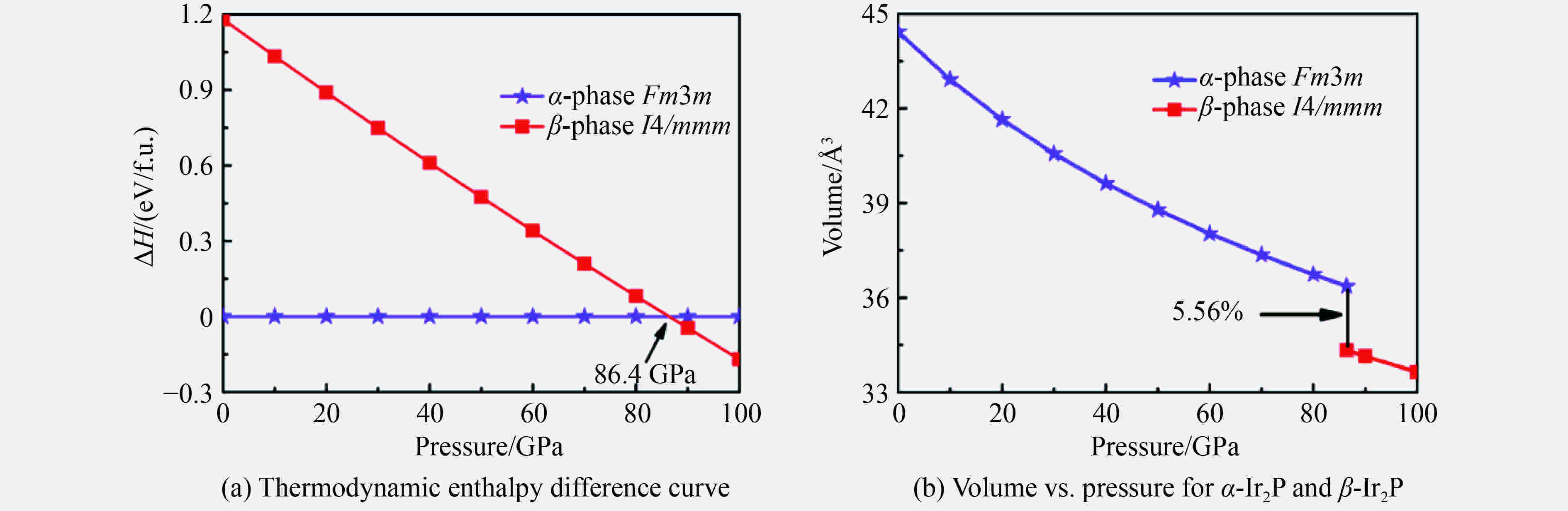
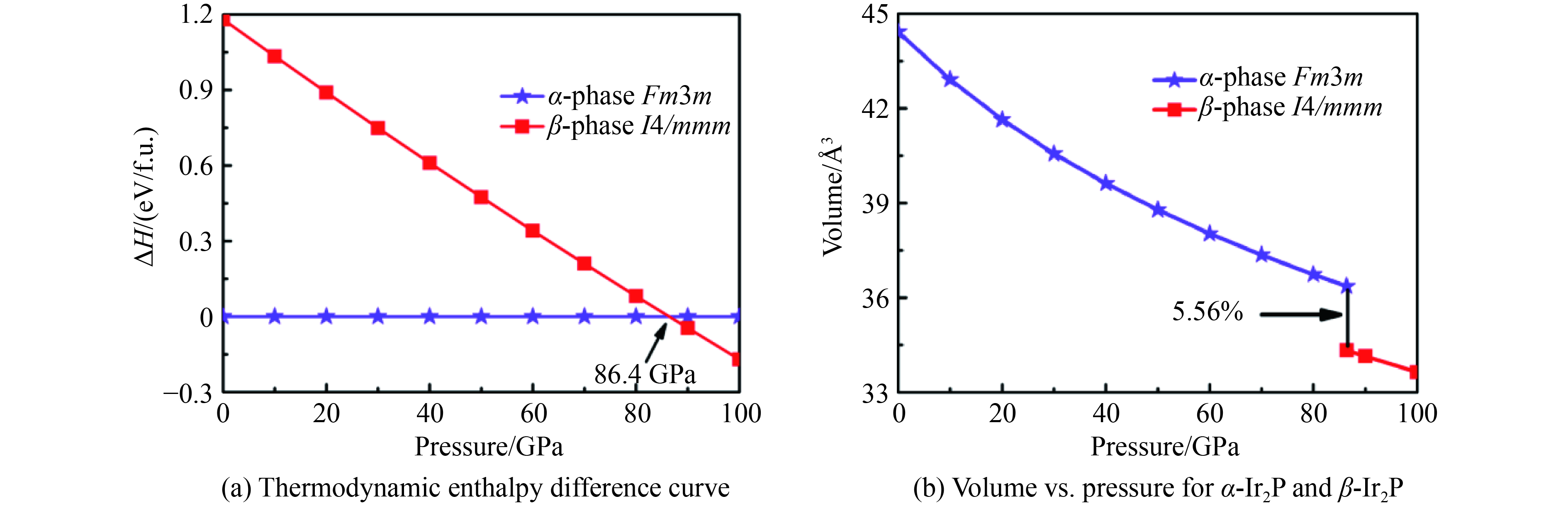
图 1 Ir2P的焓差曲线以及α-Ir2P相和β-Ir2P相体积随压强变化曲线
Figure 1. Calculated enthalpies per formula unit (f.u.) of pressure with respect to α-Ir2P and the calculated pressure versus volume phase diagram of α-Ir2P and β-Ir2P

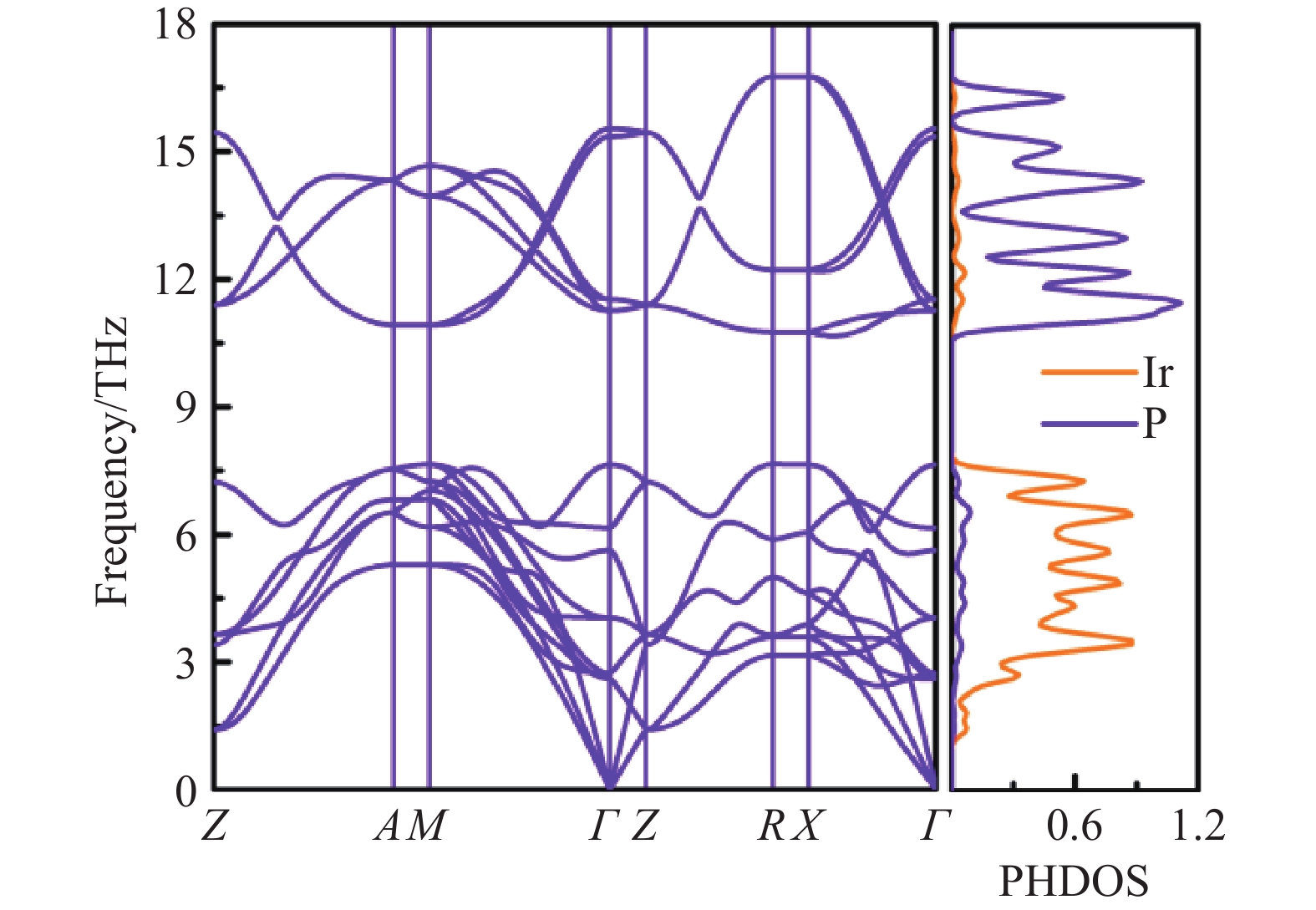
图 3 86.4 GPa时β-Ir2P相的声子谱和声子态密度
Figure 3. Phonon-dispersion curves and the PHDOS of β-Ir2P at 86.4 GPa
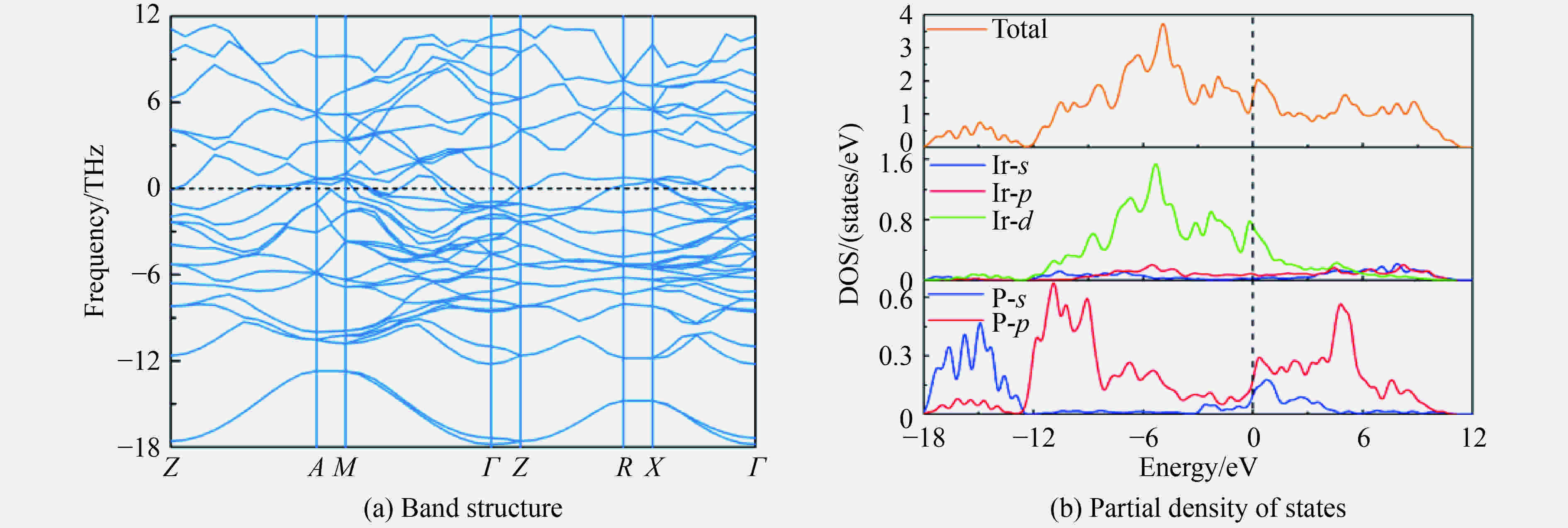
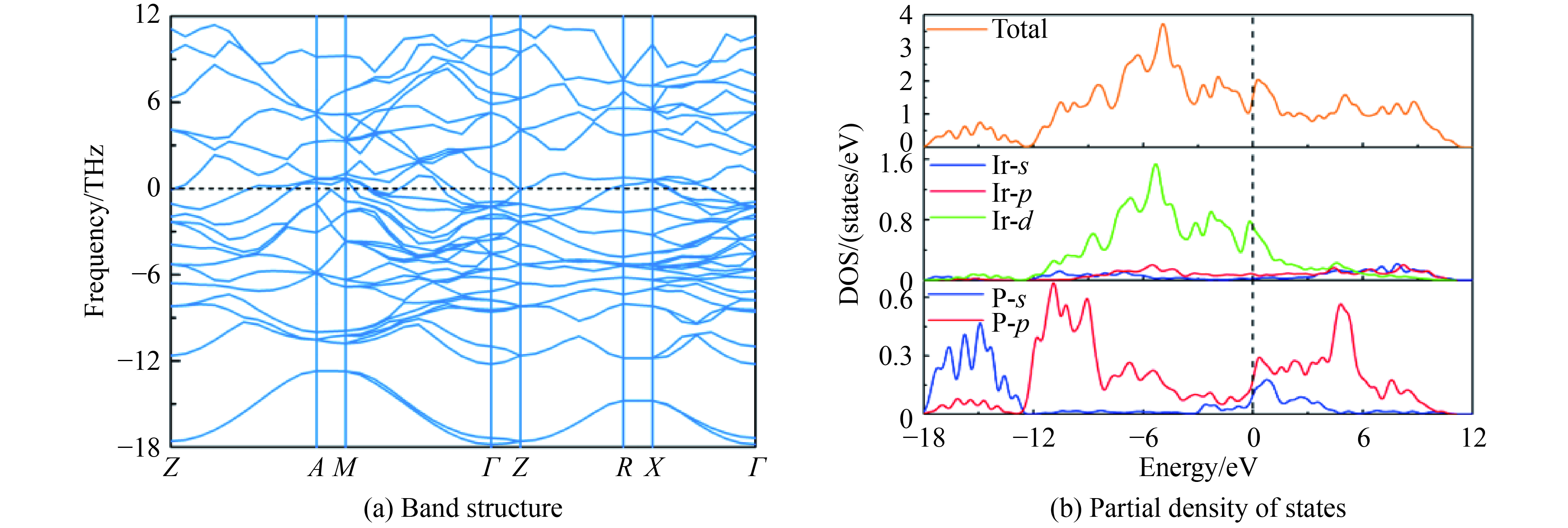
图 4 86.4 GPa下β-Ir2P相的能带结构和电子态密度
Figure 4. Band structure and partial DOS of β-Ir2P phase at 86.4 GPa

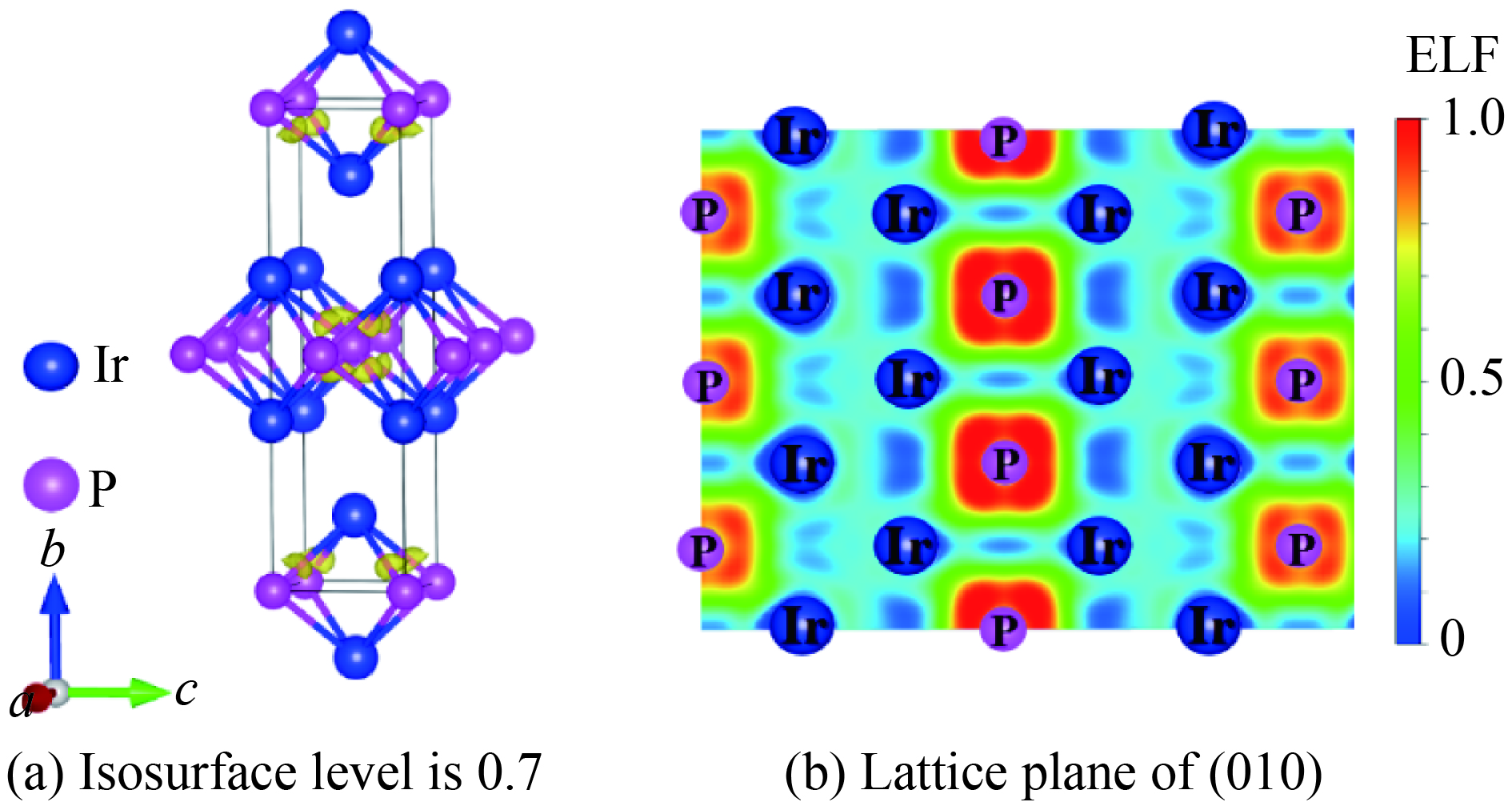
图 5 86.4 GPa下β-Ir2P相的电子局域函数
Figure 5. Electron localization function of β-Ir2P phase at 86.4 GPa
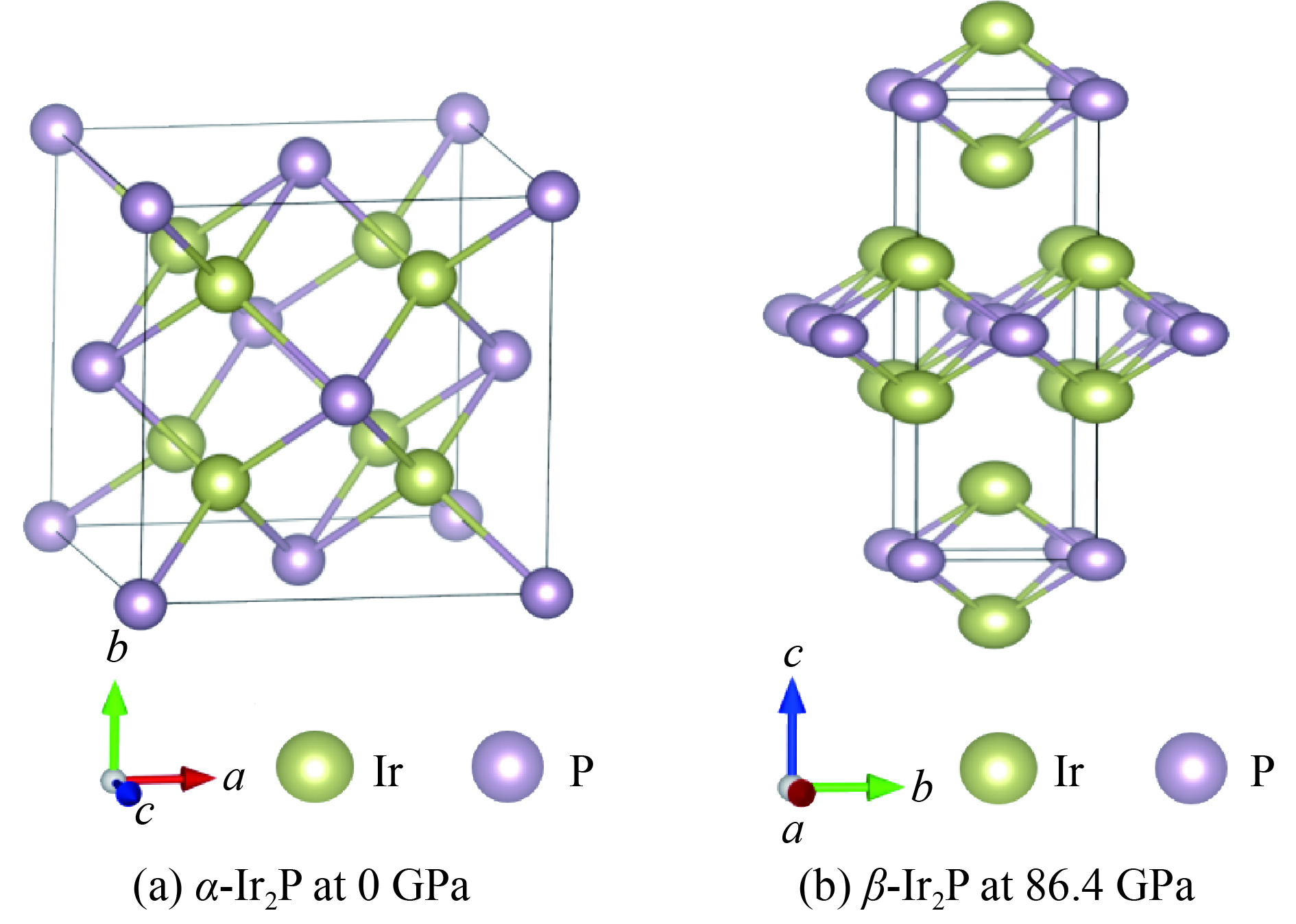
表 1 α-Ir2P相和β-Ir2P相的平衡态晶格常数和原子位置
Table 1. Lattice parameters and atomic coordinate of α-Ir2P and β-Ir2P
Phase Pressure/GPa Space group Lattice parameters Wyckoff position Atoms Site α-Ir2P 0 Fm3m a=5.622 Å(5.535 Å*), b=c=5.622 Å Ir1 8c(0.250, 0.250, 0.250) α=β=γ=90.0° P1 4a(0, 0, 0) β-Ir2P 86.4 I4/mmm a=b=2.694 Å, c=9.461 Å Ir1 4e(0.500, 0.500, 0.146) α=β=γ=90.0° P1 2a(0.500, 0.500, 0.500) Note: The asterisk represents the experimental data from Ref. [13].  下载: 导出CSV
下载: 导出CSV
表 2 86.4 GPa下β-Ir2P相的Bader电荷转移
Table 2. Calculated Bader charges of β-Ir2P phase at 86.4 GPa
Space group Pressure/GPa Atom Number Charge value/e Charge transfer/e I4/mmm 86.4 GPa Ir 2 9.19 –0.19 P 1 4.62 0.38
下载: 导出CSV
-
[1] HENKES A E, VASQUEZ Y, SCHAAK R E. Converting metals into phosphides: a general strategy for the synthesis of metal phosphide nanocrystals [J]. Journal of the American Chemical Society, 2007, 129(7): 1896–1897. doi: 10.1021/ja068502l [2] MAUVERNAY B, DOUBLET M L, MONCONDUIT L. Redox mechanism in the binary transition metal phosphide Cu3P [J]. Journal of Physics and Chemistry of Solids, 2006, 67(5/6): 1252–1257. [3] BROCK S L, SENEVIRATHNE K. Recent developments in synthetic approaches to transition metal phosphide nanoparticles for magnetic and catalytic applications [J]. Journal of Solid State Chemistry, 2008, 181(7): 1552–1559. doi: 10.1016/j.jssc.2008.03.012 [4] OYAMA S T, GOTT T, ZHAO H, et al. Transition metal phosphide hydroprocessing catalysts: a review [J]. Catalysis Today, 2009, 143(1/2): 94–107. [5] HALL J W, MEMBRENO N, WU J, et al. Low-temperature synthesis of amorphous FeP2 and its use as anodes for Li ion batteries [J]. Journal of the American Chemical Society, 2012, 134(12): 5532–5535. doi: 10.1021/ja301173q [6] BILTZ W, WEIBKE F, MAY E, et al. Alloyability of platinum and phosphorus [J]. Zeitschrift fur Anorganische und Allgemeine Chemie, 1935, 223(2): 129–143. doi: 10.1002/zaac.v223:2 [7] ZHANG X, QIN J, SUN X, et al. First-principles structural design of superhard material of ZrB4 [J]. Physical Chemistry Chemical Physics, 2013, 15(48): 20894–20899. doi: 10.1039/c3cp53893a [8] KANER R B, GILMAN J J, TOLBERT S H. Designing superhard materials [J]. Science, 2005, 308(5726): 1268–1269. doi: 10.1126/science.1109830 [9] SHI Y, ZHANG B. Correction: Recent advances in transition metal phosphide nanomaterials: synthesis and applications in hydrogen evolution reaction [J]. Chemical Society Reviews, 2016, 45(6): 1781–1781. doi: 10.1039/C6CS90013E [10] CARENCO S, PORTEHAULT D, BOISSIERE C, et al. Nanoscaled metal borides and phosphides: recent developments and perspectives [J]. Chemical Reviews, 2013, 113(10): 7981–8065. doi: 10.1021/cr400020d [11] LI W, DHANDAPANI B, OYAMA S T. Molybdenum phosphide: a novel catalyst for hydrodenitrogenation [J]. Chemistry Letters, 1998, 27(3): 207–208. doi: 10.1246/cl.1998.207 [12] OYAMA S T, GOTT T, ZHAO H, et al. Transition metal phosphide hydroprocessing catalysts: a review [J]. Catalysis Today, 2009, 143(1/2): 94–107. [13] ZUMBUSCH M. Über die strukturen des uransubsulfids und der subphosphide des iridiums und rhodiums [J]. Zeitschrift für Anorganische und Allgemeine Chemie, 1940, 243(4): 322–329. doi: 10.1002/zaac.19402430403 [14] RUNDQVIST S. Phosphides of the platinum metals [J]. Nature, 1960, 185(4705): 31. doi: 10.1038/185031a0 [15] RAUB C J, ZACHARIASEN W H, GEBALLE T H, et al. Superconductivity of some new Pt-metal compounds [J]. Journal of Physics and Chemistry of Solids, 1963, 24(9): 1093–1100. doi: 10.1016/0022-3697(63)90022-2 [16] WANG P, WANG Y, WANG L, et al. Elastic, magnetic and electronic properties of iridium phosphide Ir2P [J]. Scientific Reports, 2016, 6(9): 21787. [17] SUN X W, BIOUD N, FU Z J, et al. High-pressure elastic properties of cubic Ir2P from ab initio calculations [J]. Physics Letters A, 2016, 380(43): 3672–3677. doi: 10.1016/j.physleta.2016.08.048 [18] LIU Z J, SONG T, SUN X W, et al. Thermal expansion, heat capacity and Grüneisen parameter of iridium phosphide Ir2P from quasi-harmonic Debye model [J]. Solid State Communications, 2017, 253: 19–23. doi: 10.1016/j.ssc.2017.01.028 [19] WANG Y, LV J, ZHU L, et al. Crystal structure prediction via particle-swarm optimization [J]. Physical Review B, 2010, 82(9): 094116. doi: 10.1103/PhysRevB.82.094116 [20] MONKHORST H J, PACK J D. Special points for Brillouin-zone integrations [J]. Physical Review B, 1976, 13(12): 5188. doi: 10.1103/PhysRevB.13.5188 [21] PERDEW J P, BURKE K, ERNZERHOF M. Generalized gradient approximation made simple [J]. Physical Review Letters, 1996, 77(18): 3865–3868. doi: 10.1103/PhysRevLett.77.3865 [22] BORN M, HUANG K. Dynamical theory of crystal lattices [J]. American Journal of Physics, 1954, 39(2): 113–127. [23] SAVIN A, NESPER R, WENGERT S, et al. ELF: the electron localization function [J]. Angewandte Chemie International Edition in English, 1997, 36(17): 1808–1832. doi: 10.1002/(ISSN)1521-3773 -

 下载:
下载:

 点击查看大图
点击查看大图
计量
- 文章访问数: 7261
- HTML全文浏览量: 3161
- PDF下载量: 53
